

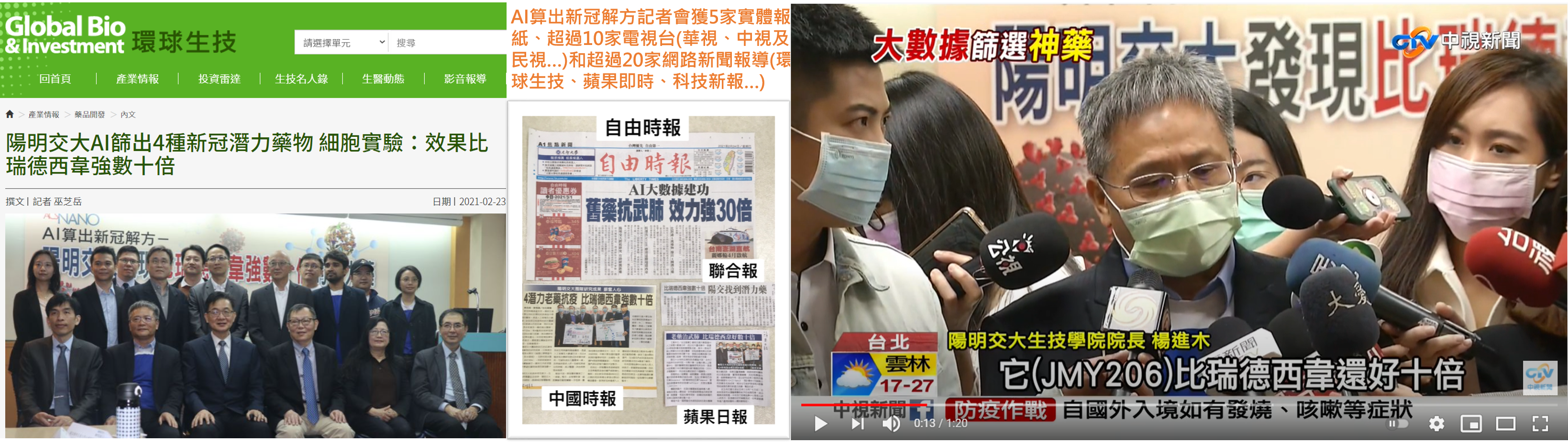
| 最新消息Latest News | 亮點成就Highlight Achievement | |
+Read more |
+Read more |
| 學術與獲獎 Academic Achievements & Awards |
產學與專利 Industry-Academia Collaboration & Patents |
國際交流與人才培育 International Exchange & Talent Education |
||
+Read more |
+Read more |
+Read more |
教授
 |
楊進木 (Jinn-Moon Yang) Dean College of Biological Science and Technology Professor Drug Design and Systems Biology Laboratory Department of Biological Science and Technology Institute of Bioinformatics & Systems Biology, National Yang Ming Chiao Tung University Address: 75 Po-Ai Street, Hsinchu, Taiwan, 30068 Office & Lab : 415-1 and 404 (Lab) in Jan Qi Biomedical Engineering Building Tel: 886-3-5712121 ext.56942; Fax: 886-3-5729288 E-mail: moon@nycu.edu.tw |
Research Overview
We have extensively explored new models of computational systems biology and computer-aided drug design to study fundamental theories of molecular interactions (e.g., Molecular Interaction Family) in cells. Our goal is to study drug-protein-pathway-cellular processes-disease relationships, and to establish the links from basic research to translational medicine. To achieve these goals, our team has been committed to integrate artificial intelligence, big data, physico-chemical concepts, and biology for exploring relationships among "molecular interactions", "cell behavior" and "drug discovery principles". To validate our models, we have rigorously collaborated with biological/clinical teams and their bioassay/clinical results support and demonstrate that our models are able to address the challenges and achieve our goals. We have continuously collaborated with researchers from various universities and institutes like NHRI, Academic Sinica, research hospitals (e.g., NTUH, TMU, and KMU) and international institutes (e.g., Kyoto University and Harvard University) for resolving unmet clinical needs and important biological issues.
Achievement
- According to Essential Science Indicators (ESI), I have three highly cited papers
- Published over 100 papers and I am the corresponding authors for some top journals as follows:
(1) Genome Biology (IF=18.011)
(2) 10 papers in Nucleic Acids Research (IF=19.16)
(3) Nature Communications (IF=17.694)
(4) ACS Nano (IF=18.027)
(5) Clinical Infectious Diseases (IF=20.999) - We have over 10 patents and discovered more than 30 lead drugs
- My h-index is 36 and the number of citations is over 5,300 (Google scholar)
Awards
- 2022: 獲科技部傑出研究獎
- 2021: 獲第18屆國家新創獎-學研新創獎
- 2021: 獲第十九屆有庠科技講座-生技醫藥類
- 2021: 獲第三十屆王民寧獎-藥學類傑出貢獻獎
- 2020: 獲中國工程師學會-傑出工程教授獎,獲總統接見。
- 2020: 獲科技部「未來科技突破獎」及「最佳媒體關注獎」
- 2018: 獲國衛院三次執行整合性醫藥衛生科技研究計畫獎牌
- 2015: 獲科技科技部「績優產學小聯盟」

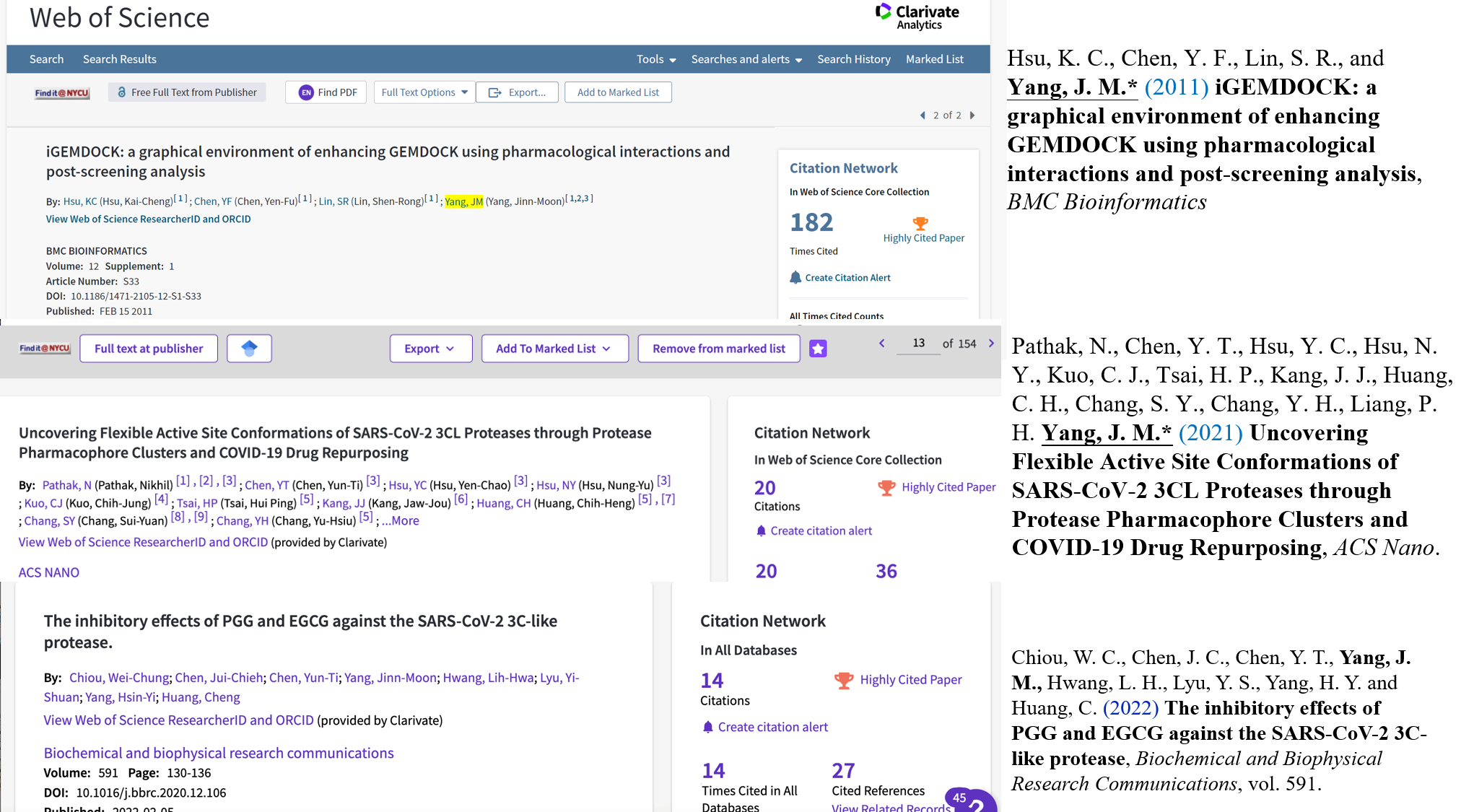
- We published Taiwan's first molecular docking paper in top-rank journal and developed it as iGEMDOCK (BMC Bioinformatics, selected as highly cited paper), which is one of widely used docking tools in the world. iGEMDOCK has been downloaded more than 100,000 times and has won the National Innovation Award. Furthermore, aiming to solve the low accuracy of virtual drug screening, we have proposed homopharma (protein-ligand interface family) and SiMMap to greatly improve the accuracy, hit rate of drug discovery, and for investigating drug binding and inhibition mechanisms supported by experimental verification. These results have been published in Genome Biology and Nucleic Acids Research, and we have obtained a US patent.
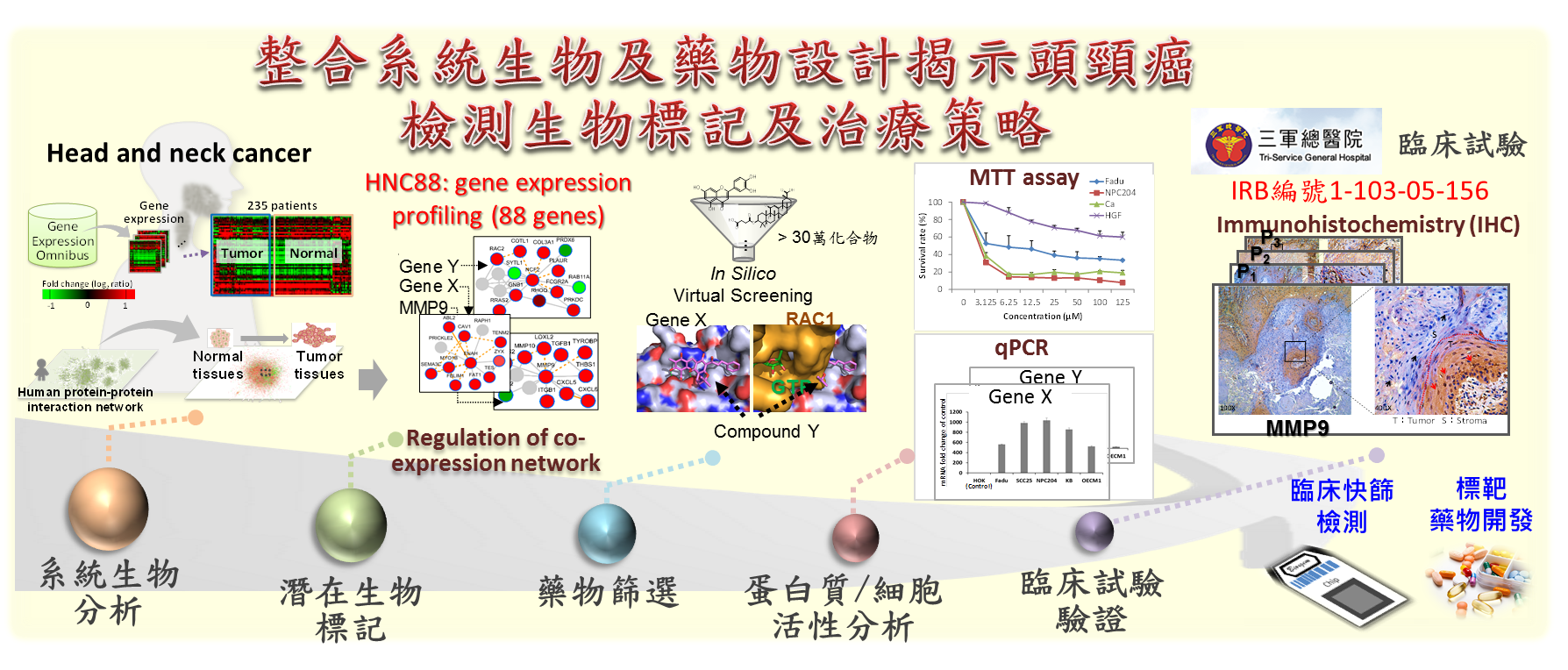
- In computational systems biology, we pioneered innovative theories and tools of protein-protein interaction family and protein module family, and published five papers in Nucleic Acids Research. Through interdisciplinary cooperation and experimental validation, we have successfully discovered biomarkers and identified disease subtypes for the diagnosis and treatment of several cancers (e.g., head and neck cancer and triple-negative breast cancer). Some of these results have been published in Nature Communications (2019) and Cancers (2020).
- For cancer precision medicine, we established Cancer Membrane Protein-regulated Networks (CaMPNets) comprising of nearly 2,000 membrane proteins (MP) in 15 diverse cancers to explore their roles in cancers, and then applied CaMPNets to clinical prognostic biomarkers identification and drug development. The cell and animal results validated our models, discovered the target MP and its FDA drug in triple-negative breast cancer. These results were published in Nature Communications (2019) and we obtained two ROC patents (2021) and filed a U.S. patent application.
- For COVID-19 drug repurposing, we have integrated pharmacophore-based models and site-moiety maps to uncover flexible active site conformations of SARS-CoV-2 3CL Proteases. Our model discovered Boceprevir (IC50=1.42 μM and EC50=49.89 μM) and Telaprevir (both HCV drugs) and Nelfinavir (HIV drug) as potential COVID-19 drugs (ACS Nano, 2021). Furthermore, we have developed a NetPharma framework, combining homopharma and evolution-based systems biology, to discover a multi-target FDA drug (JMY206, better than Remdesivir) of COVID-19. JMY206 in infected hamster model (validation by Taiwan's only P4 laboratory, National Defense Medical Center) can prevent lung damage and inflammation by modulating neutrophil infiltration and innate/ adaptive immune responses. Based on these results, we prepared papers for publication, applied two U.S. provisional patent applications (ID: 63160507 and 63160518) and clinical applications.
發表
Selected Papers
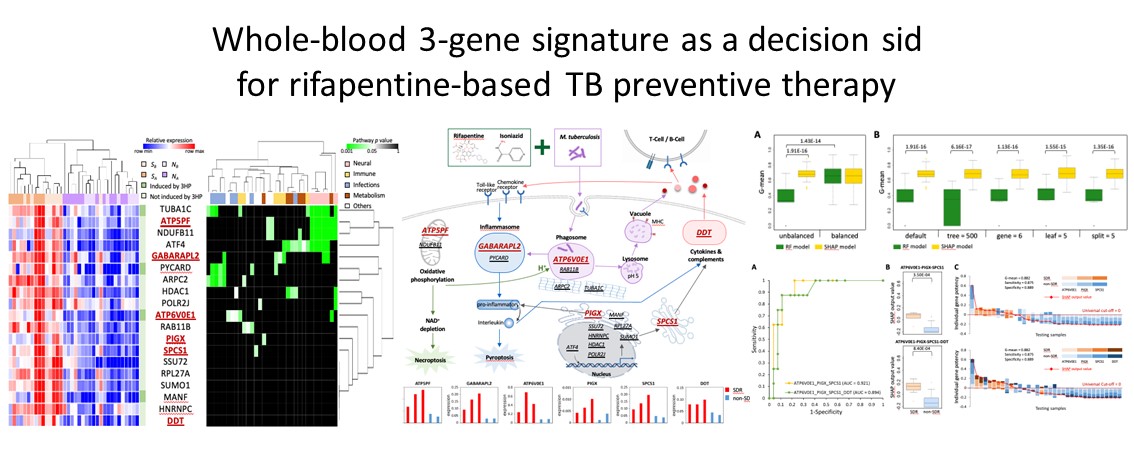
- Huang, H. L., Lee, J. Y., Lo, Y. S., Liu, I. H., Huang, S. H., Huang, Y. W., Lee, M. R., Lee, C. H., Cheng, M. H., Lu, P. L., Wang, J. Y.*, Yang, J. M.*, Chong, I.W. (2022) Whole-blood 3-gene signature as a decision sid for rifapentine-based TB preventive therapy, Clinical Infectious Diseases, doi: 10.1093/cid/ciac003. (IF=20.999)
- Huang, Y. W., Hsu, Y. C., Chuang, Y. H., Chen, Y. T., Lin, X. Y., Fan, Y. W., Pathak, N., Yang, J. M.* (2022) Discovery of moiety preference by Shapley value in protein kinase family using random forest models, BMC Bioinformatics, 23, doi: 10.1186/s12859-022-04663-5.
- Pathak, N., Chen, Y. T., Hsu, Y. C., Hsu, N. Y., Kuo, C. J., Tsai, H. P., Kang, J. J., Huang, C. H., Chang, S. Y., Chang, Y. H., Liang, P. H. and Yang, J. M.* (2020) Uncovering Flexible Active Site Conformations of SARS-CoV-2 3CL Proteases through Protease Pharmacophore Clusters and COVID-19 Drug Repurposing, ACS nano, 15(1), 857-872, doi: 10.1021/acsnano.0c07383.
- Chuang, Y. H., Huang, S. H., Hung, T. M., Lin, X. Y., Lee, J. Y., Lai, W. S. and Yang, J. M.* (2021) Convolutional neural network for human cancer types prediction by integrating protein interaction networks and omics data, Scientific Reports, 11:20691.
- Lin, C. Y., Lee, C. H., Chuang, Y. H., Lee, J. Y., Chiu, Y. Y., Lee, Y. H. W., Jong, Y. J., Hwang, J. K., Huang, S. H., Chen, L. C., Wu, C. H., Tu, S. H., Ho, Y. S., Yang, J. M.* (2019) Membrane protein-regulated networks across human cancers, Nature Communications, 10(1):3131.
- Chiu, Y. Y., Lin, C. T., Huang, J. W., Hsu, K. C., Tseng, J. H., You, S. R., and Yang, J. M.* (2013) KIDFamMap: a database of kinase-inhibitor-disease family maps for kinase inhibitor selectivity and binding mechanisms, Nucleic Acids Research, 41(Database issue):D430-40.
- Hsu, K. C., Cheng, W. C., Chen, Y. F., Wang, W. C., and Yang, J. M.* (2013) Pathway-based screening strategy for multitarget inhibitors of diverse proteins in metabolic pathways, PLOS Computational Biology, 9(7):e1003127.
- Lin, C. Y., Lin, Y. W., Yu, S. W., Lo, Y. S., and Yang, J. M.* (2012) MoNetFamily: a web server to infer homologous modules and module-module interaction networks in vertebrates, Nucleic Acids Research, 40(Web Server issue):W263-70.
- Chu, C. H., Wang, L. Y., Hsu, K. C., Chen, C. C., Cheng, H. H., Wang, S. M., Wu, C. M., Chen, T. J., Li, L. T., Liu, R. Hung, C. L., Yang, J. M.*, Kung, H. J.*, and Wang, W. C.* (2014) KDM4B as a target for prostate cancer: structural analysis and selective inhibition by a novel inhibitor, Journal of Medicinal Chemistry, 57(14):5975-85.
- Hsu, K. C., Chen, Y. F., Lin, S. R., and Yang, J. M.* (2011) iGEMDOCK: a graphical environment of enhancing GEMDOCK using pharmacological interactions and post-screening analysis, BMC Bioinformatics, 12 Suppl 1:S33.
- Tung, C. H., Huang, J. W., and Yang, J. M.* (2007) Kappa-alpha plot derived structural alphabet and BLOSUM-like substitution matrix for rapid search of protein structure database, Genome Biology, 8(3):R31.
- Chen, Y. C., Lo, Y. S., Hsu, W. C., and Yang, J. M.* (2007) 3D-partner: a web server to infer interacting partners and binding models, Nucleic Acids Research, 35(Web Server issue):W561-7.
- Chen, C. C., Hwang, J. K., and Yang, J. M.* (2006) (PS)(2): protein structure prediction server, Nucleic Acids Research, 34(Web Server issue):W152–7.
- Yang, J. M.*, Chen, Y. F., Shen, T. W., Kristal, B. S., and Hsu, D. F. (2005) Consensus scoring criteria for improving enrichment in virtual screening, Journal of Chemical Information and Modeling, 45(4):1134-46.
- Yang, J. M.*, and Chen, C. C. (2004) GEMDOCK: A generic evolutionary method for molecular docking, Proteins-Structure Function and Bioinformatics, 55(2):288-304.
Complete Journal Papers
- Chuang, Y. H., Lin, C. Y., Lee, J. C., Lee, C. H., Liu, C. L., Huang, S. H., Lee, J. Y., Lai, W. S. and Yang, J. M.* (2023) Identification of the HNSC88 Molecular Signature for Predicting Subtypes of Head and Neck Cancer, International Journal of Molecular Sciences 24: 13068.
- Huang, H. L., Luo, Y. C., Lu, P. L., Huang, C. H., Lin, K. D., Lee, M. R., Cheng, M. H., Yeh, Y. T., Kao, C. Y., Wang, J. Y., Yang, J. M.* and Chong, I. W. (2023) Gut microbiota composition can reflect immune responses of latent tuberculosis infection in patients with poorly controlled diabetes, Respiratory Research 24: 11.
- Chen, Y. T., Li, J., Chang, J. N., Luo, Y. C., Yu, W., Chen, L. C. and Yang, J. M.* (2023) Transcriptomic analysis of World Trade Center particulate Matter-induced pulmonary inflammation and drug treatments, Environment International 108027.
- Chen, Y. T., Chang, Y. H., Pathak, N., Tzou, S. C., Luo, Y. C., Hsu, Y. C., Li, T. N., Lee, J. Y., Chen, Y. C., Huang, Y. W., Yang, H. J., Hsu, N. Y., Tsai, H. P., Chang, T. Y., Hsu, S. C., Liu, P. C., Chin, Y. F., Lin, W. C., Yang, C. M., Wu, H. L., Lee, C. Y., Hsu, H. L., Liu, Y. C., Chu, J. W., Wang, L. H. C., Wang, J. Y., Huang, C. H., Lin, C. H., Hsieh, P. S., Wu Lee, Y. H., Hung, Y. J. and Yang, J. M.* (2022) Methotrexate inhibition of SARS-CoV-2 entry, infection and inflammation revealed by bioinformatics approach and a hamster model, Frontiers in Immunology 13: 7590.
- Huang, H. C., Chen, Y. T., Lin, H. H., Li, Z. Q., Yang, J. M. and Tzou, S. C. (2022) Inhibition of IRAK1 Is an Effective Therapy for Autoimmune Hypophysitis in Mice, International Journal of Molecular Sciences 23.23: 14958.
- Li, R., Lee, J. Y., Yang, J. M. and Akutsu, T. (2022) Densest subgraph-based methods for protein-protein interaction hot spot prediction, BMC Bioinformatics 23.1: 1-12.
- Li, L. Y. J., Wang, S. Yi., Yang, J. M., Chen, C. J., Tsai, C. Y., Wu, L. Y. Y., Wu, T. F., Wu, C. J. (2022) The Development and Evaluation of a Smartphone-Aided Diagnosis Application to Measure Tympanic Membrane Perforations, Ear, Nose & Throat Journal, 01455613221123361.
- Fan, Y. W., Liu, W. H., Chen, Y. T., Hsu, Y. C., Pathak, N., Huang, Y. W., and Yang, J. M.* (2022) Exploring kinase family inhibitors and their moiety preferences using deep SHapley additive exPlanations, BMC Bioinformatics, 23(4), 1-18.
- Lin, X. Y., Huang, Y. W., Fan, Y. W., Chen, Y. T., Pathak, N., Hsu, Y. C. and Yang, J. M.* (2022) Identification of pan-kinase-family inhibitors using graph convolutional networks to reveal family-sensitive pre-moieties, BMC Bioinformatics, 23(4), 1-13.
- Huang, Y. W., Hsu, Y. C., Chuang, Y. H., Chen, Y. T., Lin, X. Y., Fan, Y. W., Pathak, N. and Yang, J. M.* (2022) Discovery of moiety preference by Shapley value in protein kinase family using random forest models, BMC Bioinformatics, 23(4), 1-14.
- Wang, F., Chen, Y. T., Yang, J. M. and Akutsu, T. (2022) A novel graph convolutional neural network for predicting interaction sites on protein kinase inhibitors in phosphorylation, Scientific Reports, 12: 229.
- Huang, H. L., Lee, J. Y., Lo, Y. S., Liu, I. H., Huang, S. H., Huang, Y. W., Lee, M. R., Lee, C. H., Cheng, M. H., Lu, P. L., Wang, J. Y., Yang, J. M. and Chong, I. W. (2022) Whole-blood 3-gene Signature as a Decision Aid for Rifapentine-based TB Preventive Therapy, Clinical Infectious Diseases.
- Luo, Y. C., Huang, S. H., Pathak, N., Chuang, Y. H and Yang, J. M.* (2021) An integrated systematic approach for investigating microcurrent electrical nerve stimulation (MENS) efficacy in STZ-induced diabetes mellitus, Life Sciences, 279: 119650
- Hsu, N. Y., Pathak, N., Chen, Y. T., Hsu, Y. C. and Yang, J. M.* (2021) Pharmacophore anchor models of ATAT1 to discover potential inhibitors and lead optimization, Computational Biology and Chemistry, 93: 107513
- Chuang, Y. H., Huang, S. H., Hung, T. M., Lin, X. Y., Lee, J. Y., Lai, W. S. and Yang, J. M.* (2021) Convolutional neural network for human cancer types prediction by integrating protein interaction networks and omics data, Scientific Reports, 11: 20691.
- Chiou, W. C., Hsu, M. S., Chen, Y. T., Yang, J. M., Tsay, Y. G., Huang, H. C., and Huang, C. (2021) Repurposing existing drugs: identification of SARS-CoV-2 3C-like protease inhibitors, Journal of Enzyme Inhibition and Medicinal Chemistry, 36(1), 147-153.
- Chiou, W. C., Chen, J. C., Chen, Y. T., Yang, J. M., Hwang, L. H., Lyu, Y. S., Yang, H. Y. and Huang, C. (2021) The inhibitory effects of PGG and EGCG against the SARS-CoV-2 3C-like protease, Biochemical and biophysical research communications.
- Pathak, N., Chen, Y. T., Hsu, Y. C., Hsu, N. Y., Kuo, C. J., Tsai, H. P., Kang, J. J., Huang, C. H., Chang, S. Y., Chang, Y. H., Liang, P. H. and Yang, J. M.* (2020) Uncovering Flexible Active Site Conformations of SARS-CoV-2 3CL Proteases through Protease Pharmacophore Clusters and COVID-19 Drug Repurposing, ACS nano, 15(1), 857-872, doi: 10.1021/acsnano.0c07383.
- Hsu, K. C., HuangFu, W. C., Lin, T. E., Chao, M. W., Sung, T. Y., Chen, Y. Y., Pan, S. L., Lee, J. C., Tzou, S. C., Sun, C. M. and Yang, J. M.* (2020) A site-moiety map and virtual screening approach for discovery of novel 5-LOX inhibitors, Scientific reports, 10(1), 1-12.
- Chuang, Y. H., Lee, C. H., Lin, C. Y., Liu, C. L., Huang, S. H., Lee, J. Y., Chiu, Y. Y., Lee, J. C., Yang, J. M.* (2020) An Integrated Genomic Strategy to Identify CHRNB4 as a Diagnostic/Prognostic Biomarker for Targeted Therapy in Head and Neck Cancer, Cancers, 12(5), 1324.
- Hengphasatporn, K., Plaimas, K., Suratanee, A., Wongsriphisant, P., Yang, J. M., Shigeta, Y., Chavasiri, W., Boonyasuppayakorn, S., Rungrotmongkol, T. (2020) Target Identification Using Homopharma and Network-Based Methods for Predicting Compounds Against Dengue Virus-Infected Cells, Molecules, 25(8), 1883.
- Yang, W. Y., Rao, P. S., Luo, Y. C., Lin, H. K., Huang, S. H., Yang, J. M.*, Yuh, C. H.* (2019) Omics-based Investigation of Diet-induced Obesity Synergized with HBx, Src, and p53 Mutation Accelerating Hepatocarcinogenesis in Zebrafish Model, Cancers, 11(12), 1899.
- Simak, M., Lu, H. H. S., Yang, J. M. (2019) Boolean function network analysis of time course liver transcriptome data to reveal novel circadian transcriptional regulators in mammals, Journal of the Chinese Medical Association, 82(11), 872-880.
- Lin, C. Y., Lee, C. H., Chuang, Y. H., Lee, J. Y., Chiu, Y. Y., Lee, Y. H. W., Jong, Y. J., Hwang, J. K., Huang, S. H., Chen, L. C., Wu, C. H., Tu, S. H., Ho, Y. S., Yang, J. M.* (2019) Membrane protein-regulated networks across human cancers, Nature Communications, 10(1):3131.
- Lee, J. Y., Lin, S. Y., Lin, C. Y., Chuang, Y. H., Huang, S. H., Tseng, Y. Y., Wang, H. J., Yang, J. M.* (2019) Identification of the PCA29 gene signature as a predictor in prostate cancer, Journal of Bioinformatics and Computational Biology, 17(3):1940006.
- Lin, C. Y., Ruan, P. Y., Li, R. M., Yang, J. M., See, S., Song, J. N., Akutsu, T. (2019) Deep learning with evolutionary and genomic profiles for identifying cancer subtypes, Journal of Bioinformatics and Computational Biology, 17(3):1940005.
- Lee, C. C., Chang, W. H., Chang, Y. S., Yang, J. M., Chang, C. S., Hsu, K. C., Chen, Y. T., Liu, T. Y., Chen, Y. C., Lin, S. Y., Wu, Y. C., Chang, J. G. (2019) Alternative splicing in human cancer cells is modulated by the amiloride derivative 3,5-diamino-6-chloro-N-(N-(2,6-dichlorobenzoyl)carbamimidoyl)pyrazine-2-carboxide, Molecular Oncology, 13(8):1744-1762.
- Huang, L. C., Tam, K. W., Liu, W. N., Lin, C. Y., Hsu, K. W., Hsieh, W. S., Chi, W. M., Lee, A. W., Yang, J. M., Lin, C. L., Lee, C. H. (2018) CRISPR/Cas9 Genome Editing of Epidermal Growth Factor Receptor Sufficiently Abolished Oncogenicity in Anaplastic Thyroid Cancer, Disease Markers, 3835783.
- Huang, S. H., Lo, Y. S., Luo, Y. C., Tseng, Y. Y., Yang, J. M.* (2017) A homologous mapping method for three-dimensional reconstruction of protein networks reveals disease-associated mutations, BMC Systems Biology, 12(2):13.
- Pathak, N., Lai, M. L., Chen, W. Y., Hsieh, B. W., Yu, G. Y., Yang, J. M.* (2017) Pharmacophore anchor models of flaviviral NS3 proteases lead to drug repurposing for DENV infection, BMC Bioinformatics, 18(16):548.
- Lai, W. S., Cheng, S. Y., Lin, Y. Y., Yang, P. L., Lin, H. C., Cheng, L. H., Yang, J. M., Lee, J. C. (2017) Clinical assessment of diode laser-assisted endoscopic intrasphenoidal vidian neurectomy in the treatment of refractory rhinitis, Lasers in Medical Science, 32(9):2097-104.
- Huang, L. C., Lin, C. L., Qiu, J. Z., Lin, C. Y., Hsu, K. W., Tam, K. W., Lee, J. Y., Yang, J. M., Lee, C. H. (2017) Nicotinic Acetylcholine Receptor Subtype Alpha-9 Mediates Triple-Negative Breast Cancers Based on a Spontaneous Pulmonary Metastasis Mouse Model, Frontiers in Cellular Neuroscience, 11:336.
- Hsu, K. C., Hung, H. C., HuangFu, W. C., Sung, T. Y., Lin, T. E., Fang, M. Y., Chen, I. J., Pathak, N., Hsu, J. T. A., Yang, J. M.* (2017) Identification of neuraminidase inhibitors against dual H274Y/I222R mutant strains, Scientific Reports, 7(1):12336.
- Lin, H. C., Huan, Y. S., Chu, Y. H., Liu, S. C., Shangkuan, W. C., Lai, W. S., Yang, J. M., Lin, Y. S., Ma, K. H., Lee, J. C. (2017) Vascular anatomy is a determining factor of successful submental flap raising: a retrospective study of 70 clinical cases, PeerJ, 5:e3606.
- Hsu, H. H., Hsu, Y. C., Chang, L. J., Yang, J. M.* (2017) An integrated approach with new strategies for QSAR models and lead optimization, BMC Genomics, 18(Suppl 2):104.
- Hsu, K. C., Liu, C. Y., Lin, T. E., Hsieh, J. H., Sung, T. Y., Tseng, H. J., Yang, J. M., Huang, W. J. (2017) Novel Class IIa-Selective Histone Deacetylase Inhibitors Discovered Using an in Silico Virtual Screening Approach, Scientific Reports, 7(1):3228.
- Lai, W. S., Lin, Y. Y., Shih, C. P., Chen, H. C., Yang, P. L., Chu, Y. H., Yang, J. M., Lee, J. C. (2017) Clinical application of suction-tube-assisted septal submucosal dissection for endoscopic septoplasty, European Archives of Oto-rhino-laryngology, 274(3):1471-1475.
- Das, A., Adak, A. K., Ponnapalli, K., Lin, C. H., Hsu, K. C., Yang, J. M., Hsu, T. A., Lin, C. C. (2016) Design and synthesis of 1,2,3-triazole-containing N-acyl zanamivir analogs as potent neuraminidase inhibitors, European Journal of Medicinal Chemistry, 123:397-406.
- Huang, K. W., Hsu, K. C., Chu, L. Y., Yang, J. M., Yuan, H. S., Hsiao, Y. Y. (2016) Identification of Inhibitors for the DEDDh Family of Exonucleases and a Unique Inhibition Mechanism by Crystal Structure Analysis of CRN-4 Bound with 2-Morpholin-4-ylethanesulfonate (MES), Journal of Medicinal Chemistry, 59(17):8019-29.
- Lin, C. H., Wu, Y. R., Yang, J. M., Chen, W. L., Chao, C. Y., Chen, I. C., Lin, T. H., Wu, Y. C., Hsu, K. C., Chen, C. M., Lee, G. C., Hsieh-Li, H. M., Lee, C. M., Lee-Chen, G. J., (2016) Novel Lactulose and Melibiose Targeting Autophagy to Reduce PolyQ Aggregation in Cell Models of Spinocerebellar Ataxia 3, CNS & Neurological Disorders - Drug Targets, 15(3):351-9.
- Tsai, W. C., Yang, J. M., Liu, S. C., Chu, Y. H., Lai, W. S., Lin, Y. S., and Lee, J. C. (2015) Management of different kinds of head and neck defects with the submental flap for reconstruction, European Archives of Oto-Rhino-Laryngology, 272(12):3815-9.
- Hsu, K. C., Sung, T. Y., Lin, C. T., Chiu, Y. Y., Hsu, J. T., Hung, H. C., Sun, C. M., Barve, I., Chen, W. L., Huang, W. C., Huang, C. T., Chen, C. H., and Yang, J. M.* (2015) Anchor-based classification and type-C inhibitors for tyrosine kinases, Scientific Reports, 5:10938.
- Lee, G. C., Lin, C. H., Tao, Y. C., Yang, J. M. , Hsu, K. C., Huang, Y. J., Huang, S. H., Kung, P. J., Chen, W. L., Wang, C. M., Wu, Y. R., Chen, C. M., Lin, J. Y., Hsieh-Li, H. M., and Lee-Chen, G. J. (2015) The potential of lactulose and melibiose, two novel trehalase-indigestible and autophagy-inducing disaccharides, for polyQ-mediated neurodegenerative disease treatment, Neurotoxicology, 48:120-30.
- Chao, J. I., Wang, S. P., Wu, Y. H., Yang, J. M., Chen, CPA (2015) A novel EGFR inhibitor SP101 blocks survivin and tumor growth in the human non-small cell lung carcinoma, FASEB Journal, vol. 29 no. 1 Supplement LB113.
- Lin, C. Y., Lee, T. L., Chiu, Y. Y., Lin, Y. W., Lo, Y. S., Lin, C. T., and Yang, J. M.* (2015) Module organization and variance in protein-protein interaction networks, Scientific Reports, 5:9386.
- Lee, J. C., Lai, W. S., Ju, D. T., Chu, Y. H., and Yang, J. M. (2015) Diode laser assisted minimal invasive sphenoidotomy for endoscopic transphenoidal pituitary surgery: Our technique and results, Lasers in Surgery and Medicine, 47(3):239-42.
- Lo, Y. S., Huang, S. H., Luo, Y. C., Lin, C. Y., and Yang, J. M.* (2015) Reconstructing genome-wide protein-protein interaction networks using multiple strategies with homologous mapping, Plos One, 10(1):e0116347.
- Chiu, Y. Y., Tseng, J. H., Liu, K. H., Lin, C. T., Hsu, K. C. and Yang, J. M.* (2014) Homopharma: A new concept for exploring the molecular binding mechanisms and drug repurposing, BMC Genomics, 15 Suppl 9:S8. (Best paper award in The 13th International Conference on Bioinformatics)
- Lee, C. C., Maestre‐Reyna, M., Hsu, K. C., Wang, H. C., Liu, C. I., Jeng, W. Y., Lin, L. L., Wood, R., Chou, C. C., Yang, J. M., and Wang, A. H. (2014) Crowning Proteins: Modulating the Protein Surface Properties using Crown Ethers, Angewandte Chemie International Edition, 53(48):13054-8.
- Ma, N., Lai, C.-S., Chung, C.-H., Yang, J.-M., Hsu, K.-C., Chen, C.-Y., Chung, T.-S., Li, S., Ho, C.-T., and Pan, M.-H. (2014) 5-Demethyltangeretin is more potent than tangeretin in inhibiting dimethylbenz(a)anthracene (DMBA)/12-O-tetradecanoylphorbol-13-acetate (TPA)-induced skin tumorigenesis, Journal of Functional Foods, 11:528-37.
- Chu, C. H., Wang, L. Y., Hsu, K. C., Chen, C. C., Cheng, H. H., Wang, S. M., Wu, C. M., Chen, T. J., Li, L. T., Liu, R. Hung, C. L., Yang, J. M.*, Kung, H. J.*, and Wang, W. C.* (2014) KDM4B as a target for prostate cancer: structural analysis and selective inhibition by a novel inhibitor, Journal of Medicinal Chemistry, 57(14):5975-85.
- Wang, H. C., Ho, C. H., Hsu, K. C., Yang, J. M. and Wang, A.H. (2014) DNA mimic proteins: functions, structures, and bioinformatic analysis, Biochemistry, 53(18):2865-74.
- Wang, H. C., Hsu, K. C., Yang, J. M.Wu, M. L., Ko, T. P., Lin, S. R. and Wang, A. H. (2014) Staphylococcus aureus protein SAUGI acts as a uracil-DNA glycosylase inhibitor, Nucleic Acids Research, 42(2):1354-64.
- Liu, I. H., Lo, Y. S., and Yang, J. M.* (2013) Genome-wide structural modelling of TCR-pMHC interactions, BMC Genomics, 14 Suppl 5:S5.
- Chen, S. H., Lin, S. W., Lin, S. R., Liang, P. H., and Yang, J. M.* (2013) Moiety-Linkage Map Reveals Selective Nonbisphosphonate Inhibitors of Human Geranylgeranyl Diphosphate Synthase, Journal of Chemical Information and Modeling, 53(9):2299-311.
- Hsu, K. C., Cheng, W. C., Chen, Y. F., Wang, W. C., and Yang, J. M.* (2013) Pathway-based screening strategy for multitarget inhibitors of diverse proteins in metabolic pathways, PLOS Computational Biology, 9(7):e1003127.
- Chen, Y. J., Kay, N., Yang, J. M., Lin, C. T., Chang, H. L., Wu, Y. C., Fu, C. F., Chang, Y., Lo, S., Hou, M. F., Lee, Y. C., Hsieh, Y. C., and Yuan, S. S. (2013) Total Synthetic Protoapigenone WYC02 Inhibits Cervical Cancer Cell Proliferation and Tumour Growth through PIK3 Signalling Pathway, Basic & Clinical Pharmacology & Toxicology, 113(1):8-18.
- Hsu, K. C., Hung, H. C., Horng, J. T., Fang, M. Y., Chang, C. Y., Li, L. T., Chen, I. J., Chen, Y. C., Chou, D. L., Chang, C. W., Hsieh, H. P., Yang, J. M.*, and J. T., Hsu*. (2013) Parallel screening of wild-type and drug-resistant targets for anti-resistance neuraminidase inhibitors, PLoS ONE, 8(2):e56704.
- Lin, C. Y., Chen, Y. C., Lo, Y. S., and Yang, J. M.* (2013) Inferring homologous protein-protein interactions through pair position specific scoring matrix, BMC Bioinformatics, 14 Suppl 2:S11.
- Liu, I. H., Lo, Y. S. and Yang, J. M.* (2013) Template-based scoring functions for visualising biological insights of H-2K(b)-peptide-TCR complexes, International Journal of Data Mining and Bioinformatics, 8(3):326-37.
- Lin, C. H., Chang, T. C., Das, A., Fang, M. Y., Hung, H. C., Hsu, K. C., Yang, J. M., M, v. I., Mong, K. K., Hsu, T. A., and Lin, C. C. (2013) Synthesis of acylguanidine zanamivir derivatives as neuraminidase inhibitors and the evaluation of their bio-activities, Organic & Biomolecular Chemistry, 11(24):3943-8.
- Chiu, Y. Y., Lin, C. T., Huang, J. W., Hsu, K. C., Tseng, J. H., You, S. R., and Yang, J. M.* (2013) KIDFamMap: a database of kinase-inhibitor-disease family maps for kinase inhibitor selectivity and binding mechanisms, Nucleic Acids Research, 41(Database issue):D430-40.
- Chiu, Y. Y., Lin, C. Y., Lin, C. T., Hsu, K. C., Chang, L. Z., and Yang, J. M.* (2012) Space-related pharma-motifs for fast search protein binding motifs and polypharmacological targets, BMC Genomics, 13 Suppl 7:S21.
- Huang, J. W., Lin, W. F., and Yang, J. M.* (2012) Antigenic sites of H1N1 influenza virus hemagglutinin revealed by natural isolates and inhibition assays, Vaccine, 30(44):6327-37.
- Lin, C. Y., Lin, Y. W., Yu, S. W., Lo, Y. S., and Yang, J. M.* (2012) MoNetFamily: a web server to infer homologous modules and module-module interaction networks in vertebrates, Nucleic Acids Research, 40(Web Server issue):W263-70.
- Hsu, S. C., Chang, C. P., Tsai, C. Y., Hsieh, S. H., Wu-Hsieh, B. A., Lo, Y. S., and Yang, J. M. (2012) Steric recognition of T-cell receptor contact residues is required to map mutant epitopes by immunoinformatical programmes, Immunology, 136(2):139-52.
- Chang, K. M., Chen, S. H., Kuo, C. J., Chang, C. K., Guo, R. T., Yang, J. M., and Liang, P. H. (2012) Roles of amino acids in the Escherichia coli octaprenyl diphosphate synthase active site probed by structure-guided site-directed mutagenesis, Biochemistry, 51(16):3412-9.
- Cheng, W. C., Chen, Y. F., Wang, H. J., Hsu, K. C., Lin, S. C., Chen, T. J., Yang, J. M.*, and Wang, W. C. (2012) Structures of Helicobacter pylori shikimate kinase reveal a selective inhibitor-induced-fit mechanism, PLoS ONE, 7(3):e33481.
- Hsu, K. C., Cheng, W. C., Chen, Y. F., Wang, H. J., Li, L. T., Wang, W. C., and Yang, J. M.* (2012) Core site-moiety maps reveal inhibitors and binding mechanisms of orthologous proteins by screening compound libraries, PLoS ONE, 7(2):e32142.
- Hsu, K. C., Chen, Y. F., and Yang, J. M.* (2012) GemAffinity: a scoring function for predicting binding affinity and virtual screening, International Journal of Data Mining and Bioinformatics, 6(1):27-41.
- Clinciu, D. L., Yang, J. M., Hsu, K. C., Lo, C. C., Wallace, S., and Yu, H. C. (2011) The relevance of protein-ligand interaction profiles in computer-aided novel compound design and applications, Current Bioinformatics, 6(3):383-8.
- Liu, I. H., Lo, Y. S., and Yang, J. M.* (2011) PAComplex: a web server to infer peptide antigen families and binding models from TCR-pMHC complexes, Nucleic Acids Research, 39(Web Server issue):W254-60.
- Tsai, C. C., Liu, H. F., Hsu, K. C.,Yang, J. M., Chen, C. P., Liu, K. K., Hsu, T. S., and Chao, J. I. (2011) 7-Chloro-6-piperidin-1-yl-quinoline-5,8-dione (PT-262), a novel ROCK inhibitor blocks cytoskeleton function and cell migration, Biochemical Pharmacology, 81(7):856-65.
- Hsu, K. C., Chen, Y. F., Lin, S. R., and Yang, J. M.* (2011) iGEMDOCK: a graphical environment of enhancing GEMDOCK using pharmacological interactions and post-screening analysis, BMC Bioinformatics, 12 Suppl 1:S33.
- Huang, J. W., and Yang, J. M.* (2011) Changed epitopes drive the antigenic drift for influenza A (H3N2) viruses, BMC Bioinformatics, 12 Suppl 1:S31.
- Clinciu, D. L., Chen, Y. F., Ko, C. N., Lo, C. C., and Yang, J. M.* (2010) TSCC: Two-Stage Combinatorial Clustering for virtual screening using protein-ligand interactions and physicochemical features, BMC Genomics, 11 Suppl 4:S26.
- Lo, Y. S., Chen, Y. C., and Yang, J. M.* (2010) 3D-interologs: an evolution database of physical protein- protein interactions across multiple genomes, BMC Genomics, 11 Suppl 3:S7.
- Chen, Y. F., Hsu, K. C., Lin, S. R., Wang, W. C., Huang, Y. C., and Yang, J. M.* (2010) SiMMap: a web server for inferring site-moiety map to recognize interaction preferences between protein pockets and compound moieties, Nucleic Acids Research, 38(Web Server issue):W424-30.
- Lo, Y. S., Lin, C. Y., and Yang, J. M.* (2010) PCFamily: a web server for searching homologous protein complexes, Nucleic Acids Research, 38(Web Server issue):W516-22.
- Liu, K. P., Hsu, K. C., Huang, J. W., Chang, L. S., and Yang, J. M.* (2010) ATRIPPI: An atom-residue preference scoring function for protein-protein interactions, International Journal on Artificial Intelligence Tools, 19(3):251-66.
- Chin, K. H., Lee, Y. C., Tu, Z. L., Chen, C. H., Tseng, Y. H., Yang, J. M.*, Ryan, R. P., McCarthy, Y., Dow, J. M., Wang, A. H. J., and Chou, S. H. (2010) The c-AMP receptor-like protein clp is a novel c-di-gmp receptor linking cell-cell signaling to virulence gene expression in xanthomonas campestris, Journal of Molecular Biology, 396(3):646-62.
- Chen, C. C., Hwang, J. K., and Yang, J. M.* (2009) (PS)(2)-v2: template-based protein structure prediction server, BMC Bioinformatics, 10:366.
- Lin, F. K., Pan, C. L., Yang, J. M., Chuang, T. J., and Chen, F. C. (2009) CAPIH: A web interface for comparative analyses and visualization of host-HIV protein-protein interactions, BMC Microbiology, 9:164
- Chen, C. C., Lin, C. Y., Lo, Y. S., and Yang, J. M.* (2009) PPISearch: a web server for searching homologous protein-protein interactions across multiple species, Nucleic Acids Research, 37(Web Server issue):W369-75.
- Hung, H. C., Tseng, C. P., Yang, J. M., Ju, Y. W., Tseng, S. N., Chen, Y. F., Chao, Y. S., Hsieh, H. P., Shih, S. R., and Hsu, J. T. A. (2009) Aurintricarboxylic acid inhibits influenza virus neuraminidase, Antiviral Research, 81(2):123-31.
- Huang, J. W., King, C. C., and Yang, J. M.* (2009) Co-evolution positions and rules for antigenic variants of human influenza A/H3N2 viruses, BMC Bioinformatics, 10 Suppl 1:S41.
- Yao, Y. Y., Shrestha, K. L., Wu, Y. J., Tasi, H. J., Chen, C. C., Yang, J. M., Ando, A., Cheng, C. Y., and Li, Y. K. (2008) Structural simulation and protein engineering to convert an endo-chitosanase to an exo-chitosanase, Protein Engineering Design & Selection, 21(9):561-6.
- Yang, M. C., Guan, H. H., Yang, J. M., Ko, C. N., Liu, M. Y., Lin, Y. H., Huang, Y. C., Chen, C. J., and Mao, S. J. T. (2008) Rational design for crystallization of beta-lactoglobulin and Vitamin D(3) complex: revealing a secondary binding site, Crystal Growth & Design, 8(12):4268-76.
- Yang, M. C., Guan, H. H., Liu, M. Y., Lin, Y. H.,Yang, J. M., Chen, W. L., Chen, C. J., and Mao, S. J. T. (2008) Crystal structure of a secondary vitamin D(3) binding site of milk beta-lactoglobulin, Proteins-Structure Function and Bioinformatics, 71(3):1197-210.
- Yang, C. Y., Chang, C. H., Yu, Y. L., Lin, T. C. E., Lee, S. A., Yen, C. C., Yang, J. M., Lai, J. M., Hong, Y. R., Tseng, T. L., Chao, K. M., and Huang, C. Y. F. (2008) PhosphoPOINT: a comprehensive human kinase interactome and phospho-protein database, Bioinformatics, 24(16):i14-20.
- Chiu, Y. Y., Hwang, J. K., and Yang, J. M.* (2008) Soft energy function and generic evolutionary method for discriminating native from nonnative protein conformations, Journal of Computational Chemistry, 29(9):1364-73.
- Chang, Y. L., Tsai, H. K., Kao, C. Y., Chen, Y. C., Hu, Y. J., and Yang, J. M.* (2008) Evolutionary conservation of DNA-contact residues in DNA-binding domains, BMC Bioinformatics, 9 Suppl 6:S3.
- Yang, J. M., Chen, Y. F., Tu, Y. Y., Yen, K. R., and Yang, Y. L. (2007) Combinatorial computational approaches to identify tetracycline derivatives as flavivirus inhibitors, PLoS ONE, 2(5):e428.
- Tung, C. H., and Yang, J. M.* (2007) fastSCOP: a fast web server for recognizing protein structural domains and SCOP superfamilies, Nucleic Acids Research, 35(Web Server issue):W438-43.
- Tung, C. H., Huang, J. W., and Yang, J. M.* (2007) Kappa-alpha plot derived structural alphabet and BLOSUM-like substitution matrix for rapid search of protein structure database, Genome Biology, 8(3):R31.
- Chen, Y. C., Lo, Y. S., Hsu, W. C., and Yang, J. M.* (2007) 3D-partner: a web server to infer interacting partners and binding models, Nucleic Acids Research, 35(Web Server issue):W561-7.
- Yang, J. M.*, and Tung, C. H. (2006) Protein structure database search and evolutionary classification, Nucleic Acids Research, 34(13):3646-59.
- Chen, C. C., Hwang, J. K., and Yang, J. M.* (2006) (PS)(2): protein structure prediction server, Nucleic Acids Research, 34(Web Server issue):W152–7.
- Yang, J. M.*, and Shen, T. W. (2005) A pharmacophore-based evolutionary approach for screening selective estrogen receptor modulators, Proteins-Structure Function and Bioinformatics, 59(2):205-20.
- Yang, J. M.*, Chen, Y. F., Shen, T. W., Kristal, B. S., and Hsu, D. F. (2005) Consensus scoring criteria for improving enrichment in virtual screening, Journal of Chemical Information and Modeling, 45(4):1134-46.
- Yang, J. M.*, and Chen, C. C. (2004) GEMDOCK: A generic evolutionary method for molecular docking, Proteins-Structure Function and Bioinformatics, 55(2):288-304.
- Yang, J. M.* (2004) Development and evaluation of a generic evolutionary method for protein-ligand docking, Journal of Computational Chemistry, 25(6):843-57.
- Tsai, H. K., Yang, J. M., Tsai, Y. F., and Kao, C. Y. (2004) Some issues of designing genetic algorithms for traveling salesman problems, Soft Computing, 8(10):689-97.
- Tsai, H. K., Yang, J. M., Tsai, Y. F., and Kao, C. Y. (2004) An evolutionary algorithm for large traveling salesman problems, IEEE Transactions on Systems Man and Cybernetics Part B-Cybernetics, 34(4):1718-29.
- Tsai, H. K., Yang, J. M.*, Tsai, Y. F., and Kao, C. Y. (2004) An evolutionary approach for gene expression patterns, IEEE Transactions on Information Technology in Biomedicine, 8(2):69-78.
- Yu, C. S., Wang, J. Y., Yang, J. M., Lyu, P. C., Lin, C. J., and Hwang, J. K. (2003) Fine-grained protein fold assignment by support vector machines using generalized npeptide coding schemes and jury voting from multiple-parameter sets, Proteins-Structure Function and Bioinformatics, 50(4):531-6.
- Tsai, H. K., Yang, J. M., Tsai, Y. F., and Kao, C. Y. (2003) Heterogeneous selection genetic algorithms for traveling salesman problems, Engineering Optimization, 35(3):297-311.
- Lin, E. S., Yang, J. M., and Yang, Y. S. (2003) Modeling the binding and inhibition mechanism of nucleotides and sulfotransferase using molecular docking, Journal of the Chinese Chemical Society, 50(3B):655-63.
- Chuang, C. C., Chen, C. Y., Yang, J. M., Lyu, P. C., and Hwang, J. K. (2003) Relationship between protein structures and disulfide bonding patterns, Proteins-Structure Function and Genetics, 53(1):1-5.
- Chu, Y. W., and Yang, J. M.* (2003) Finding regularity in various types of secondary protein structures, Journal of Information Science and Engineering, 19:943-952.
- Yang, J. M.*, Tsai, C. H., Hwang, M. J., Tsai, H. K., Hwang, J. K., and Kao, C. Y. (2002) GEM: A Gaussian evolutionary method for predicting protein side-chain conformations, Protein Science, 11(8):1897-907.
- Yang, J. M.*, Lin, C. J., and Kao, C. Y. (2002) A robust evolutionary algorithm for global optimization, Engineering Optimization, 34(5):405-425.
- Yang, J. M.*, and Kao, C. Y. (2001) Efficient evolutionary algorithm for the thin-film synthesis of inhomogeneous optical coatings, Applied Optics, 40(19):3256-67.
- Yang, J. M.*, and Kao, C. Y. (2001) An evolutionary algorithm for the synthesis of multilayer coatings at oblique light incidence, Journal of Lightwave Technology, 19(4):559-70.
- Yang, J. M.*, and Kao, C. Y. (2001) A robust evolutionary algorithm for training neural networks, Neural Computing & Applications, 10(3):214-30.
- Yang, J. M.*, Horng, J. T., Lin, C. J., and Kao, C. Y. (2001) Optical coating designs using the family competition evolutionary algorithm, Evolutionary Computation, 9(4):421-43.
- Yang, J. M.*, and Kao, C. Y. (2000) Integrating adaptive mutations and family competition into genetic algorithms as function optimizer, Soft Computing, 4(2):89-102.
- Yang, J. M.*, and Kao, C. Y. (2000) A family competition evolutionary algorithm for automated docking of flexible ligands to proteins, IEEE Transactions on Information Technology in Biomedicine, 4(3):225-37.
- Yang, J. M.*, and Kao, C. Y. (2000) Flexible ligand docking using a robust evolutionary algorithm, Journal of Computational Chemistry, 21(11):988-998.
- Yang, J. M.*, Horng, J. T., and Kao, C. Y. (2000) A genetic algorithm with adaptive mutations and family competition for training neural networks, International Journal of Neural Systems, 10(5):333-52.
Patents
- Yang, J. M., Hsieh, P. S., Huang, C. H., Chang, Y. H., Pathak, N., Hsu, Y. C., Chen, Y. T., Hsu, N. Y., METHOD AGAINST SEVERE ACUTE RESPIRATORY SYNDROME CORONAVIRUS 2 INFECTION. US11786530B2, 2023/10/17
- 楊進木、謝博軒、黃志恒、張聿秀、倪 齊歐、許彥超、陳筠媞、許農育。對抗嚴重急性呼吸道症候群冠狀病毒2 型感染的方法。中華民國專利:發明第I815341號,2023/09/11。
- 楊進木、李嘉華、李容羽、陳筠媞。羅索沙星在抑制三陰性乳癌的轉移上的用途。中華民國專利:發明第I804455號,2023/06/01。
- 楊進木、林峻宇、邱一原、李容羽、何元順、李嘉華。安非他酮和醫藥組合物在製備治療癌症之藥物的用途及抑制腫瘤細胞遷移的方法。中華民國專利:發明第I736452號,2021/08/11。
- Yang, J. M., Hsu, K. C., Sung T. Y., Lin S. R., Wang Y. M., Hsu, K. M., Lin, H. P., and Liu, W. C., Selective inhibitors for protein kinases and pharmaceutical composition and use thereof. US20160096848A1, 2016/04/07
- Chao, J. I., Wang, S. P., Chen, C. P., Yin, K. H., Yang, J. M., and Wu, Y. H., Compound for promoting apoptosis of cancer cells and a pharmaceutical composition containing the same and uses thereof. US20160068495A1, 2016/03/10
- 楊進木、林峻宇、邱一原、李容羽、何元順、李嘉華。安非他酮和醫藥組合物在製備治療癌症之藥物的用途及抑制腫瘤細胞遷移的方法。中華民國專利:發明第 I713875號,2020/12/21。
- 楊進木、許凱程、宋姿瑩、林伸融、王雲銘、許光美、林欣平、劉婉婷。蛋白質激酶之選擇性抑制劑、其醫藥組成物及其用途。中華民國專利:發明第 I538914 號,2016/06/21。
- 楊進木、許凱程、黃兆祺、陳文亮、趙瑞益、黃文傑。一種專一性表皮生長因子接受器之選擇性抑制劑、其醫藥組成物及其用途。中華民國專利:發明第 I536987 號,2016/06/11。
- Wang, W. C., Cheng, W. C., and Yang, J. M., Method of inhibiting the growth of Helicobacter pylori. US8175860B2, 2012/05/08
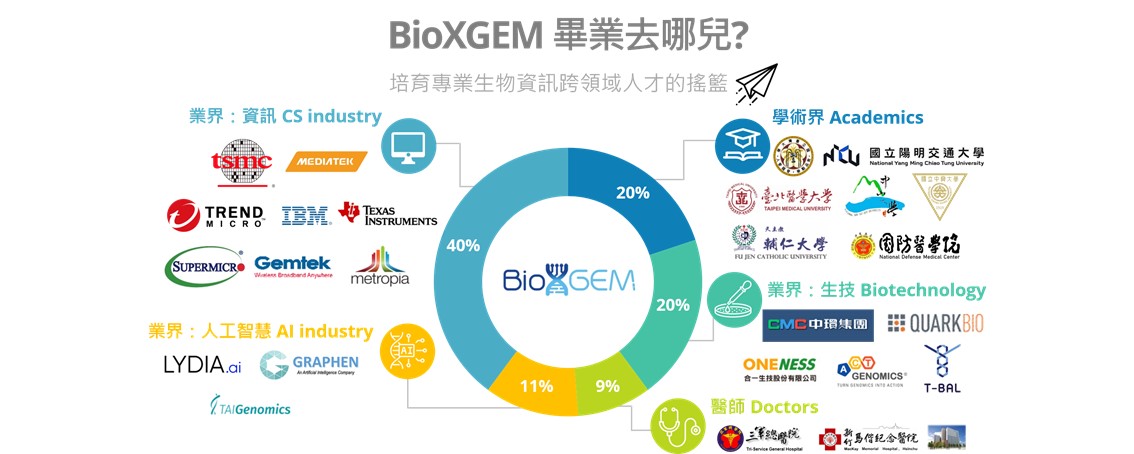
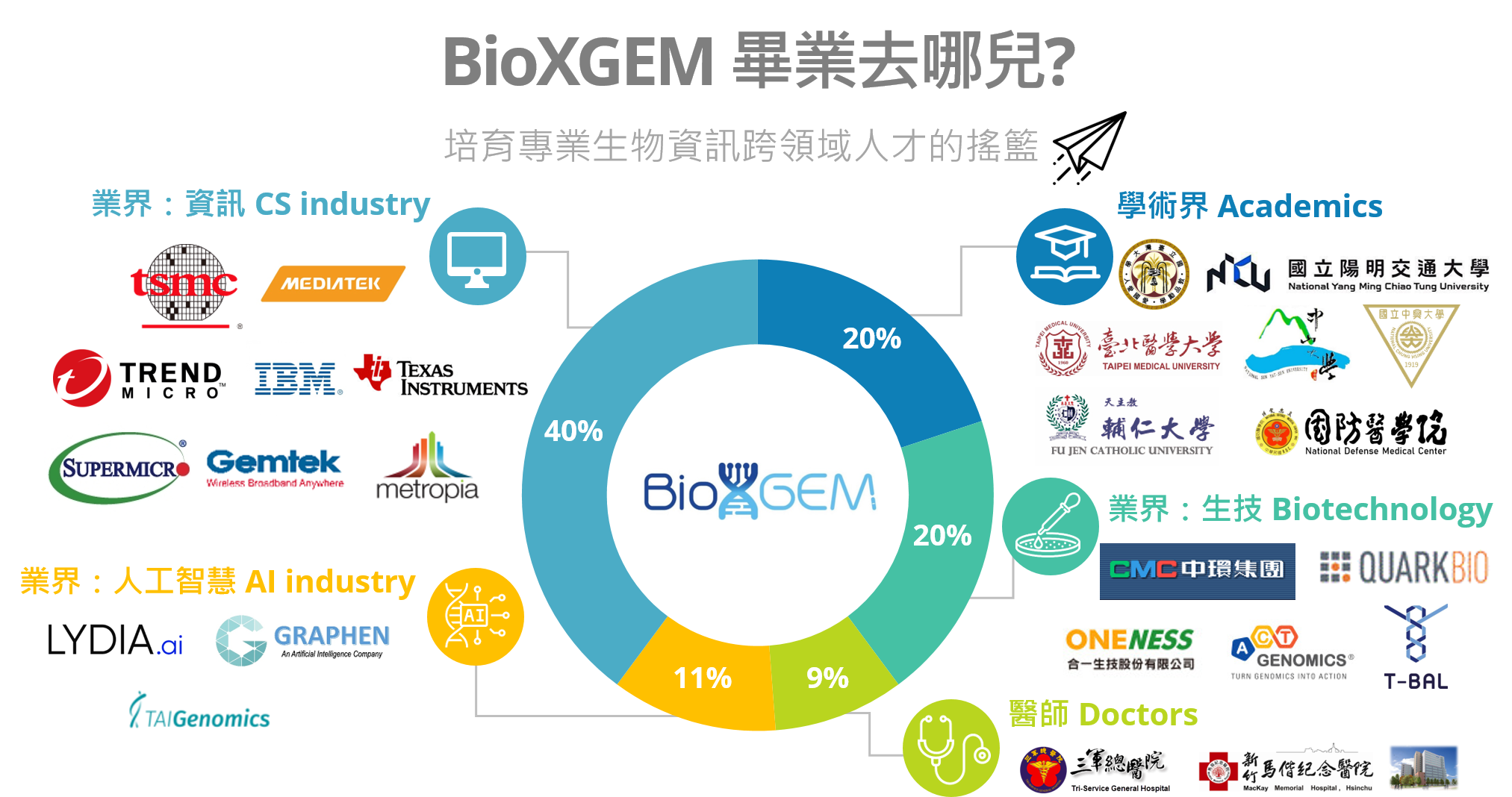
畢業生出路
博士班畢業生 (共20名)
| 姓名___________________________ | 畢業學年度_____________________ | 目前任職_______________________ |
| 莊佾軒 | 2023 | -- |
| 劉宛欣 (共同指導) | 2021 | 北一女中教師 |
| Nikhil Pathak | 2020 | 普睿思股份有限公司 |
| 史蔓莉 (共同指導) | 2018 | -- |
| 黃星翰 | 2017 | 圖策科技 |
| 陳世勳 | 2013 | 國立臺灣大學 |
| 林志達 | 2013 | 癌症顧問公司 |
| 邱一原 | 2013 | 台積電 |
| 林峻宇 | 2013 | 國立陽明交通大學 |
| 劉怡馨 | 2012 | -- |
| 羅宇書 | 2011 | 台達電子 |
| 許凱程 | 2010 | 臺北醫學大學 |
| 丹尼爾 (共同指導) | 2010 | -- |
| 黃章維 | 2010 | 奎克生技光電股份有限公司 |
| 陳志杰 (共同指導) | 2009 | 國立中山大學 |
| 陳彥甫 | 2009 | 中環股份有限公司 |
| 董其樺 | 2008 | 輔仁大學 |
| 陳俊辰 | 2008 | -- |
| 張耀霖 (共同指導) | 2008 | -- |
| 朱彥煒 (共同指導) | 2005 | 國立中興大學 |
MD. Ph.D.畢業生 (共2名)
| 姓名___________________________ | 畢業學年度_____________________ | 目前任職_______________________ |
| 賴文森 | 2017 | 國軍臺中 803 總醫院 |
| 李日清 | 2015 | 國防醫學院 三軍總醫院 |
碩士班畢業生 (共65名)
| 姓名___________________________ | 畢業學年度_____________________ | 目前任職_______________________ |
| 藍靖涵 | 2024 | Synopsys 新思科技 |
| 林韋霖 | 2024 | -- |
| 周治瑗 | 2024 | -- |
| 張振寧 | 2024 | -- |
| 楊欣儒 | 2022 | 科林研發 |
| 陳宜群 | 2021 | -- |
| 黃宇薇 | 2020 | 聯發科技 |
| 林庠豫 | 2020 | 圖策科技 |
| 范祐瑋 | 2020 | -- |
| 吳承軒 | 2019 | 慧邦科技 |
| 林楚芸 | 2019 | 尚禾亞 |
| 李可愉 | 2019 | -- |
| 蔡宜君 | 2018 | Lydia.ai |
| 劉威廷 | 2018 | 盟基生技 |
| 趙千嫣 | 2018 | 聯發科技 |
| 李力渝 | 2018 | 國立台灣大學醫學院醫學系內科 |
| 郭超望 | 2018 | chimes AI |
| 洪慈懋 | 2018 | 台智基因體股份有限公司 |
| 陳筠媞 (共同指導) | 2018 | -- |
| 林思妤 | 2016 | -- |
| 巫思穎 | 2016 | 台積電 |
| 李容羽 | 2016 | -- |
| 李哲勳 | 2016 | -- |
| 楊方琪 | 2015 | -- |
| 許元綸 | 2015 | 台灣國際商業機器股份有限公司 |
| 顏佳儀 | 2015 | 合一生技 |
| 沈培鈞 | 2014 | 台灣活性脂質股份有限公司 |
| 林傳東 | 2014 | -- |
| 陳怡君 | 2014 | 台積電 |
| 莊佾軒 | 2014 | -- |
| 曾仁琥 | 2014 | 美創資通 |
| 林昱葦 | 2014 | 趨勢科技 |
| 劉冠秀 | 2013 | Supermicro |
| 林伸融 | 2013 | -- |
| 李采凌 | 2012 | -- |
| 徐瑋澤 | 2012 | -- |
| 游尚文 | 2011 | -- |
| 李淩婷 | 2011 | 正文科技 |
| 尤宣人 | 2011 | 趨勢科技 |
| 許彥超 | 2011 | -- |
| 黃御哲 | 2010 | -- |
| 林怡瑋 | 2010 | 行動基因生技股份有限公司 |
| 林韋帆 | 2010 | -- |
| 張力仁 | 2009 | -- |
| 林峻宇 | 2009 | 國立陽明交通大學 |
| 陳彥修 | 2008 | -- |
| 劉康平 | 2008 | -- |
| 陳祐德 | 2007 | -- |
| 羅宇書 | 2007 | 台達電子 |
| 許凱程 | 2006 | 臺北醫學大學 |
| 陳右儒 | 2006 | 聯發科技 |
| 陳永強 | 2005 | 新竹馬偕醫院 |
| 楊登凱 | 2005 | -- |
| 葛振寧 | 2005 | -- |
| 黃章維 | 2005 | 奎克生技光電股份有限公司 |
| 陳宏助 | 2004 | -- |
| 張立人 | 2004 | -- |
| 林柏村 | 2004 | -- |
| 邱一原 | 2004 | 台積電 |
| 董其樺 | 2004 | 輔仁大學 |
| 林建宏 | 2003 | -- |
| 沈再威 | 2003 | -- |
| 陳彥甫 | 2003 | 中環股份有限公司 |
| 張育祥 | 2002 | -- |
成員
PI
楊進木 (Jinn-Moon Yang) |
|
|
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||
| Back to top | ||||||||||||||||||||||||||||||||||||||||||||||||||||




















Alumni
Ph.D.
| Name | Major Fields in BS | Memo (Research Groups, Research Directions, Publications) |
| 李容羽 | Animal Science | July 2023, Ph.D. Systems Biology: Protein-protein interaction |
| 莊佾軒 | Computer Science | July 2023, Ph.D. Systems Biology: Protein-protein interaction |
| Nikhil Pathak | Pharmacy | July 2020, Ph.D. Drug Design for Pathogenic and infectious Diseases - Dengue |
| 黃星翰 (Sing-Han Huang) |
Biochemical Science and Technology | October 2017, Ph.D. Human Brain Aging and Neurodegeneration Disease Journal paper: 2 papers |
| 林峻宇 (Chun-Yu Lin) |
Biological Technology | July 2014, Ph.D. Systems Biology: Protein-protein interaction Journal paper: 11 papers |
| 林志達 (Chih-Ta Lin) |
Biological Technology | July 2014, Ph.D. Anticancer drug: Kinase-specific inhibitors & networks Journal paper: 5 papers |
| 邱一原 (Yi-Yuan Chiu) |
Computer Science | July 2014, Ph.D. Computer-aided drug design Journal paper: 4 papers |
| 陳世勳 (Shih-Hsun Chen) |
Biochemical Engineering | December 2013, Ph.D. Computer-aided drug design Journal paper: 3 papers |
| 劉怡馨 (I-Hsin Liu) |
Life Science | July 2013, Ph.D. Systems Biology: Protein-MHC-TCR interaction Journal paper: 3 papers |
| 羅宇書 (Yu-Shu Lo) |
Life Science | July 2012, Ph.D. Systems Biology: Protein-protein interaction Journal paper: 10 papers |
| 許凱程 (Kai-Cheng Hsu) |
Biological Technology | July 2011, Ph.D. Computer-aided drug design Journal paper: 16 papers |
| 陳彥甫 (Yen-Fu Chen) |
Applied Chemistry | July 2010, Ph.D. Computer-aided drug design Journal paper: 6 papers (Win the 2007 National Innovation Award) |
| 黃章維 (Jhang-Wei Huang) |
Biological Technology | September 2010, Ph.D. Influenza Virus: Antigenic Drift Analysis, Antigen-Antibody interaction Journal paper: 4 papers |
| 陳俊辰 (Chun-Chen Chen) |
Biological Technology | June 2009, Ph.D. Systems Biology: Protein-protein interaction Journal paper: 3 papers |
| 董其樺 (Chi-Hua Tung) |
Biological Science | June 2009, Ph.D. Structural Bioinformatics: 3D-BLAST, Antigen-Antibody interaction Journal paper: 3 papers |
| 張耀霖 (Yao-Lin Chang) |
Computer Science | June 2008, Ph.D. Title: A 3D-regulog approach to predict protein-DNA binding partners and binding model (Co-advised student with Cheng-Yan Kao in NTU) |
| 朱彥煒 (Yen-Wei Chu) |
Computer Science | June 2006, Ph.D. Title: Finding Protein Secondary Structure Regularity and Related Applications (Co-advised student with Chuen-Tsai Sun in NCTU) |
| Back to top |
MD. Ph.D.
| Name | Major Fields in BS | Memo (Research Groups, Research Directions, Publications) |
| 賴文森 (Wen-Sen Lai) |
Department of Otolaryngology, Taichung Armed forces General Hospital | June 2018, MD. Ph.D. Journal paper: 22 papers |
| 李日清 (Jih-Chin Lee) |
Institute of Medical Sciences, The National Defense Medical Center | July 2016, MD. Ph.D. Journal paper: 41 papers |
Master
| Name | Major Fields in BS | Memo (Research Groups, Research Directions, Publications) |
| 林韋霖 Wei-Lin Lin) |
Biotechnology | March 2024, Master Computer-aided drug design |
| 藍靖涵 (Ching-Han Lan) |
Applied Bioscience Technology | March 2024, Master Computer-aided drug design |
| 周治瑗 (Chi-hyuan Chou) |
Applied Life science | January 2024, Master Systems Biology |
| 楊欣儒 (Hsin-Ju Yang) |
Food sciences | July 2022, Master Computer-aided drug design |
| 陳宜群 (Yi-Cyun Chen) |
Animal Science | August 2021, Master Computer-aided drug design |
| 黃宇薇 (Yu-Wei Huang) |
Computer Science and Information Engineering | December 2020, Master Computer-aided drug design |
| 林庠豫 (Xiang-Yu Lin) |
Biological Technology | September 2020, Master Computer-aided drug design |
| 范祐瑋 (You-Wei Fan, Ethan) |
Microbiology, Immunology and Biopharmaceuticals | September 2020, Master Computer-aided drug design |
| 吳承軒 (Cheng-Hsuan Wu) |
Biochemistry | July 2020, Master Computer-aided drug design |
| 林楚芸 (Chu-Yun Lin) |
Food and nutrition | July 2020, Master Computer-aided drug design |
| 李可愉 (Ke-Yu Lee) |
Department of Chemical Engineering | July 2019, Master Computer-aided drug design |
| 劉威廷 (Wei-Ting Liu) |
Food sciences | March 2019, Master Systems Biology |
| 郭超望 (Chao-Wang Kuo) |
Medical Technology | March 2019, Master Systems Biology |
| 李力渝 (Li-Yu Li) |
Food Science | March 2019, Master Computer-aided drug design |
| 趙千嫣 (Chien-Yen Chao) |
Computer Science and Information Engineering | March 2019, Master Computer-aided drug design |
| 洪慈懋 (Tzu-Mao Hung) |
Nutrition | March 2019, Master Computer-aided drug design |
| 蔡宜君 (Yi-Chun Tsai) |
Biological Technology | January 2019, Master Computer-aided drug design |
| 巫思穎 (Szu-Ying Wu) |
Microbiology | July 2017, Master Computer-aided drug design |
| 李容羽 (Jung-Yu Lee) |
Animal Science | July 2017, Master Systems Biology: Protein-protein interaction |
| 許元綸 (Yuan-Luen Sheu) |
Information Management | July 2016, Master Systems Biology: Protein-protein interaction |
| 顏佳儀 (Chia-Yi Yen) |
Biological Technology | July 2016, Master Computer-aided drug design |
| 楊方琪 (Yi-Jyun Jhen) |
Biological Technology | July 2016, Master Systems Biology: Protein-protein interaction |
| 李哲勳 (Jhuan-Dong Lin) |
Biological Technology | July 2016, Master Computer-aided drug design |
| 莊佾軒 (Yi-Shen Chung) |
Computer Science | July 2015, Master Systems Biology: Protein-protein interaction |
| 沈培鈞 (Pei-Jyun Chen) |
Life Science | July 2015, Master Computer-aided drug design |
| 陳怡君 (Yi-Jyun Jhen) |
Information Management | July 2015, Master Systems Biology: Protein-protein interaction |
| 林傳東 (Jhuan-Dong Lin) |
Earth Sciences | July 2015, Master Computer-aided drug design |
| 曾仁琥 (Jen-Hu Tseng) |
Pharmacy | July 2015, Master Computer-aided drug design |
| 林昱葦 (Yu-Wei Lin) |
Biochemical Technology | July 2014, Master Systems Biology: Protein-Protein interaction |
| 劉冠秀 (Kuan-Shiu Liu) |
Computer Science | July 2014, Master Computer-aided drug design |
| 李采凌 (Tsai-Ling Lee) |
Computer Science | July 2013, Master Systems Biology: Protein-protein interaction Journal paper: 1 paper |
| 徐瑋澤 (Wei-Ze Syu) |
Life Science | July 2013, Master Computer-aided drug design |
| 尤宣人 (Syuan-Ren You) |
Computer Science | July 2012, Master Computer-aided drug design |
| 游宇震 (Yu-Chen, Yu) |
Management Information System | July 2012, Master Systems Biology: Protein-protein interaction |
| 李淩婷 (Ling-Ting Li) |
Medicine Management | July 2012, Master Computer-aided drug design Title: Core site-moiety maps of histone demethylase family for multi-target inhibitors and binding mechanisms Journal paper: 3 papers |
| 林伸融 (Shen-Rong Lin) |
Computer Science | July 2014, Master Computer-aided drug design Title: iGemdock 2.0 and SiMMap: Post-screening analysis for structure-based drug design Journal paper: 4 paper |
| 黃御哲 (Yu-Chi Huang) |
Biological Technology | July 2011, Master Computer-aided drug design Title: A web server for lead optimization using site-moiety map: A case study on neuraminidase Journal paper: 1 paper |
| 林怡瑋 (Yi-Wei Lin) |
Life Science | July 2011, Master Systems Biology: Protein-protein interaction Title: Template-based Homologous Modules through Protein-protein Interaction Families Journal paper: 2 papers |
| 許彥超 (Yen-Chao Hsu) |
Computer Science | December 2011, Master Computer-aided drug design Title: Moiety-based site-moiety map |
| 林韋帆 (Wei-Fan Lin) |
Life Science | July 2011, Master Influenza Virus: Antigenic Drift Analysis Title: Genetic and antigenic analysis to the hemagglutinin of influenza A H1N1 virus and comparisons with H3N2 virusJournal paper: 1 paper |
| 張力仁 (Li-Jen Chang) |
Life Science | July 2010, Master Structural Bioinformatics |
| 林峻宇 (Chun-Yu Lin) |
Biological Technology | May 2010, Master Systems Biology: Protein-protein interaction Journal paper: 2 papers |
| 陳彥修 (Yen-Hsiu Chen) |
Life Science | June 2009, Master Systems Biology: Protein-DNA/Protein-RNA interaction |
| 劉康平 (Kang-Ping Liu) |
Computer Science | June 2009, Master Systems Biology: Protein-protein interaction Journal paper: 1 paper |
| 陳祐德 (Yu-Te Chen) |
Aquaculture | June 2008, Master Title: A New Profile Method to Predict Protein-ligand Binding Site |
| 羅宇書 (Yu-Shu Lo) |
Life Science | June 2008, Master Title: Template-Based Scoring Function for Predicting Protein-protein Interaction Journal paper: 2 papers |
| 許凱程 (Kai-Cheng Hsu) |
Biological Technology | June 2007, Master Title: Binding Affinity Analysis of Protein-ligand Complexes |
| 陳右儒 (Yo-Ru Chen) |
Computer Science | June 2007, Master Title: iGEMDOCK: A graphical-automatic drug design system for docking, screening and post-analysis (Win the 2007 National Innovation Award) |
| 楊登凱 (Teng-Kai Yang) |
Computer Science | June 2006, Master Title: Structural Binding Pocket Clustering and Protein-ligand Interaction Analysis for ATP-binding Proteins |
| 陳永強 (Yung-Chiang Chen) |
Life Science | June 2006, Master Title: Inferring Domain Annotated Protein-Protein Interactions through 3D-Domain Interologs Journal paper: 2 papers |
| 葛振寧 (Chen-Neng Ko) |
Biological Science | June 2006, Master Title: Structure-based Compound Screening for Helicobacter pylori shikimate kinase (HpSK) Journal paper: 1 paper |
| 黃章維 (Jhang-Wei Huang) |
Biological Technology | June 2006, Master Title: Predicting Antigenic Variants of Influenza A H3N2 Viruses by Building Relationships between Genetic Evolution and Antigenic Drift Journal paper: 2 papers |
| 董其樺 (Chi-Hua Tung) |
Biological Science | June 2005, Master Title: PiSA-BLAST: A New Tool for Protein Structure Alignment and Database Search (Win the prize of 2005 Best M.S. and Ph. D. Dissertation Award hosted by IICM (Institute of Information & Computing Machinery)) Website: 3D-BLAST Journal paper: 3 papers |
| 邱一原 (Yi-Yuan Chiu) |
Computer Science | June 2005, Master Title: Optimizing New Energy Functions for Protein Folding Journal paper: 1 paper |
| 林柏村 (Po-Tsun Lin) |
Life Science | June 2005, Master Title: LigSeeSVM: Support Vector Machines and Data fusion for Ligand-based Compound Screening and Applications to GPCR and GABAA |
| 陳宏助 (Heng-Chu Chen) |
Life Science | June 2005, Master Title: Inferring Protein-protein Interactions from Structural Domain-domain Interactions |
| 張立人 (Li-Jen Chang) |
Medical Technology | June 2005, Master Title: Integrating GEMDOCK with GEM-PLS and GEM-kNN for QSAR modeling of huAChE and AGHO |
| 沈再威 (Tsai-Wei Shen) |
Biological Science | June 2004, Master Title: Enhancing GEMDOCK on Virtual Database Screening and Application to Envelope Protein and D-Hydantoinase (Win the prize of 2004 Best M.S. and Ph. D. Dissertation Award hosted by IICM (Institute of Information & Computing Machinery)) Website: GEMDOCK Journal paper: 2 papers |
| 林建宏 (Chien-Heng Lin) |
Biological Science | June 2004, Master Title: MuLiSA: Analysis and Identification of Functional Motifs and Residues in Proteins by Multiple Ligand-bound Structure Alignments |
| 陳彥甫 (Yen-Fu Chen) |
Applied Chemistry | June 2004, Master Title: Validation and Application of GEMDOCK and Application of Data Fusion in Virtual Screening Journal paper: 2 papers |
| 張育祥 (Yu-Hsiang Chang) |
Life Science | June 2003, Master Title: A New Energy Model for Protein-Protein Interaction Prediction |
| Back to top |
研究及合作夥伴
建立藥物-蛋白質-生化途徑-疾病機制的關連建立基礎研究-轉譯醫學-產業的連結 |
|
|
 |
OverviewOur research focused on computer-aided drug design (GEMDOCK and SiMMap), structural bioinformatics (3D-BLAST, (PS)2 and fastSCOP), and systems biology (PPISearch and 3D-partner), especially investigating the molecular interfaces (e.g., protein-protein, protein-DNA, protein-ligand interactions). Our main contributions and innovations are described as follows: I) Discover the protein-compound interaction family for multi-target drug design and new uses for old drug [1-4]. II) The first team proposed protein-protein interactions (interface family) across multiple species [5-8]. III) We have developed the first tool, called 3D-BLAST which is as fast as BLAST and has the features of BLAST (e.g. robust statistical basis, and effective and reliable search capabilities), to search large protein structures (> 30000) within 3 seconds in the world [9-11]. |
|
 |
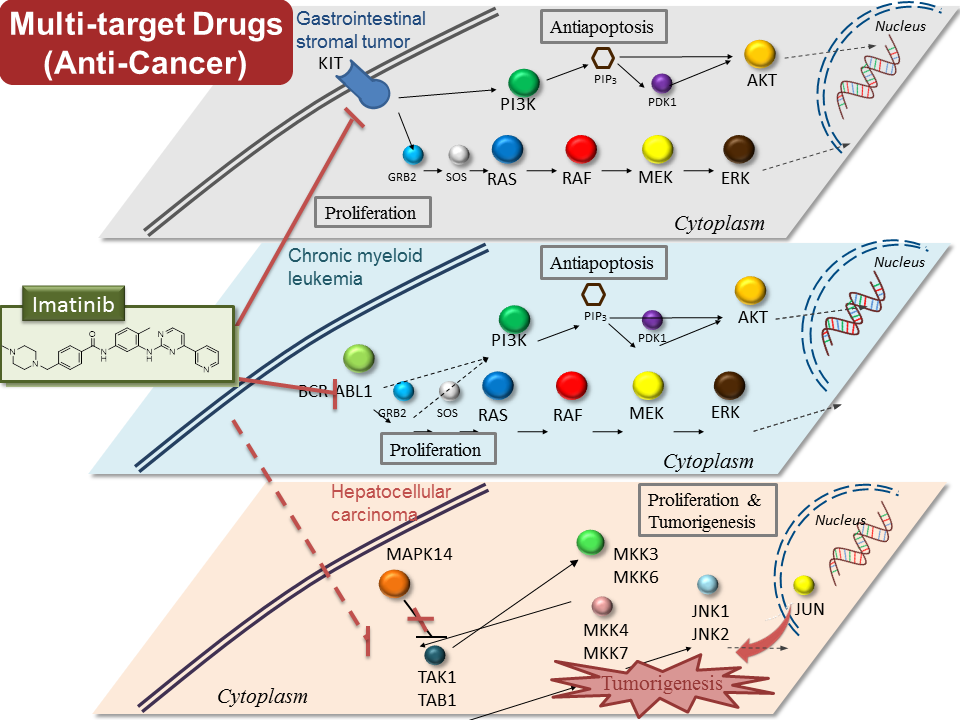 |
Drug discovery: Pharmalogs and HomopharmaFor structure-based protein-ligand interactions and drug discovery, we have developed a molecular docking tool, namely GEMDOCK [3, 4, 13, 14], which is one of wide-used tools in the world. We cooperated with 10 research teams on diverse drug targets. We cooperated with over 10 laboratories which successfully applied our tools for identifying lead compounds and protein functional sites and protein engineering. For example, GEMDOCK has been successfully applied to identify the lead compounds for dengue virus envelop protein (cooperating with Yang YL in NCTU)[2]; inhibitors of influenza virus neuraminidase (cooperating with Hsu TA in NHRI) [15]; lead compounds of ROCK kinase (cooperating with Chao JI in NCTU) for anti-cancer drugs; antibiotics of shikimate kinases of Mycobacterium tuberculosis and Helicobacter pylori (cooperating with Wang WC in NTHU). We have successfully applied our tools for discovering the secondary vitamin D3 binding site of milk beta-lactoglobulin (cooperating with Mao JI in NCTU)[16, 17]; the binding mechanism between c-di-GMP ad cAMP Receptor-Like Protein CLP (cooperating with Chao SH in NCHU) [18]. The timed citations of two related papers [3, 13] are 58 and 61, respectively, from 2004. These related papers were cited over 180 times. We got the 2007 National Innovation Award due to the achievement of iGEMDOCK.在藥物開發部份,本實驗室透過獨立研發的全台第一個藥物嵌合軟體 GEMDOCK(榮獲 2007 國家新創獎)與超過 400 個 CPU 的計算資源,可快速的針對特定藥物標靶於虛擬藥物資料庫中(超過 30 萬筆小分子化合物)進行篩選,並利用全球首創的區域官能基地圖技術分析標靶蛋白的藥物結合機制,進而篩選出新型抑制劑。有別於其他計算實驗室缺乏自主實驗驗證,我們已建構由全波長式多功能微量分析儀暨冷螢光數位影像分析系統為主的高效藥篩平台,可對篩選出的抑制劑進行活性分析,並可針對常見癌症(肝癌、乳癌、頭頸癌)或發炎反應進行細胞實驗分析。此外,我們並與淡水馬偕醫院與三軍總醫院合作進行臨床試驗,是國內少數從計算跨足轉譯醫學的藥物設計與系統生物實驗室。在癌症與許多複雜疾病的主要藥物標靶-蛋白激酶上,我們透過上述程序,發現了特定受體酪氨酸激酶專屬的結合位點(Type-C pocket),並成功針對此位點篩選出選擇性抑制劑(Type-C inhibitors),而大規模蛋白激酶抑制活性分析結果亦顯示出高選擇性,並能克服傳統化療藥物所產生的常見抗藥性突變,順利突破此類年產值高達 185 億美金的藥物目前所面對的瓶頸--選擇性與抗藥性,為其選擇性與抗藥性設計開闢另一個方向。 |
|
 |
|
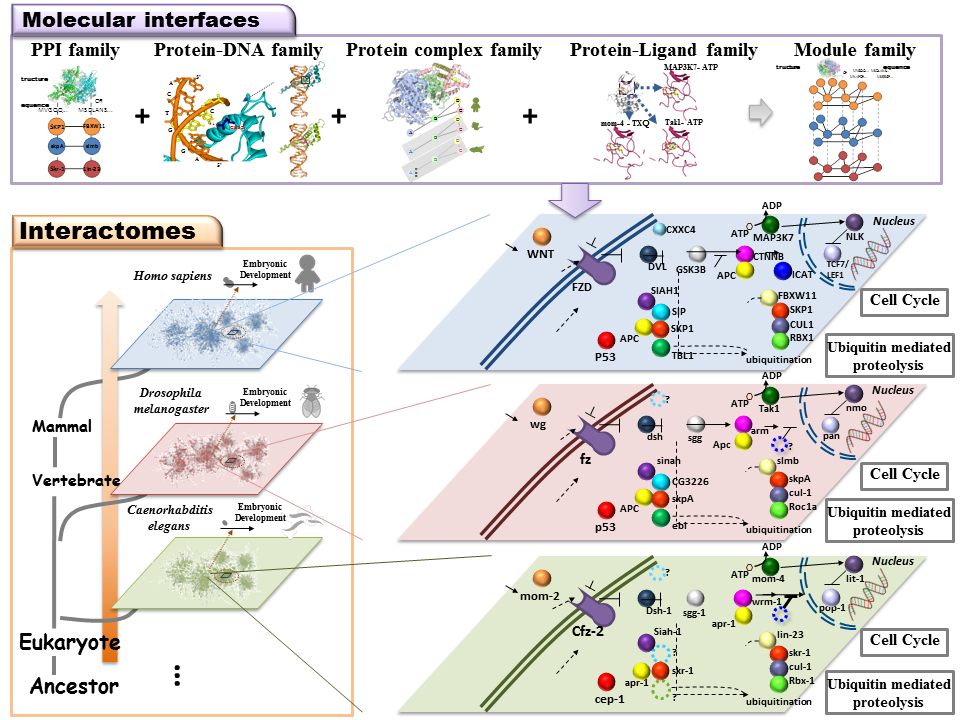
Molecular interface families (protein-drug, protein-protein, and protein-DNA)In protein-peptide interactions, we improved our PPI template-based scoring function with template similarity and interacting force to guarantee statistically significant interface similarity between peptide candidates and structure templates. Our model, considering both MHC-peptide and peptide-TCR interfaces, could provide visualization and biological insights of MHC-peptide-TCR binding models. We believe that it is very helpful for the development of peptide-based vaccines. For protein-DNA interactions, we developed a new approach, namely 3D-regulogs, to infer protein-DNA binding partners by using the concept of regulog and crystal structures of protein-DNA complex as templates [12]. This approach also provides binding models, interacting amino acids, and DNA bases of predicted partners. For protein-ligand interactions, we have introduced site-moiety maps [1], which recognize interaction preferences between protein pockets and compound moieties. SiMMap was developed to identify site-moiety maps from query protein structures and their docked (or co-crystallized) compounds. Over 1,400 users have created more than 1,500 site-moiety maps for different proteins. Site-moiety map is able to provide biological insights and useful for drug discovery and lead optimization. |
|
 |
|
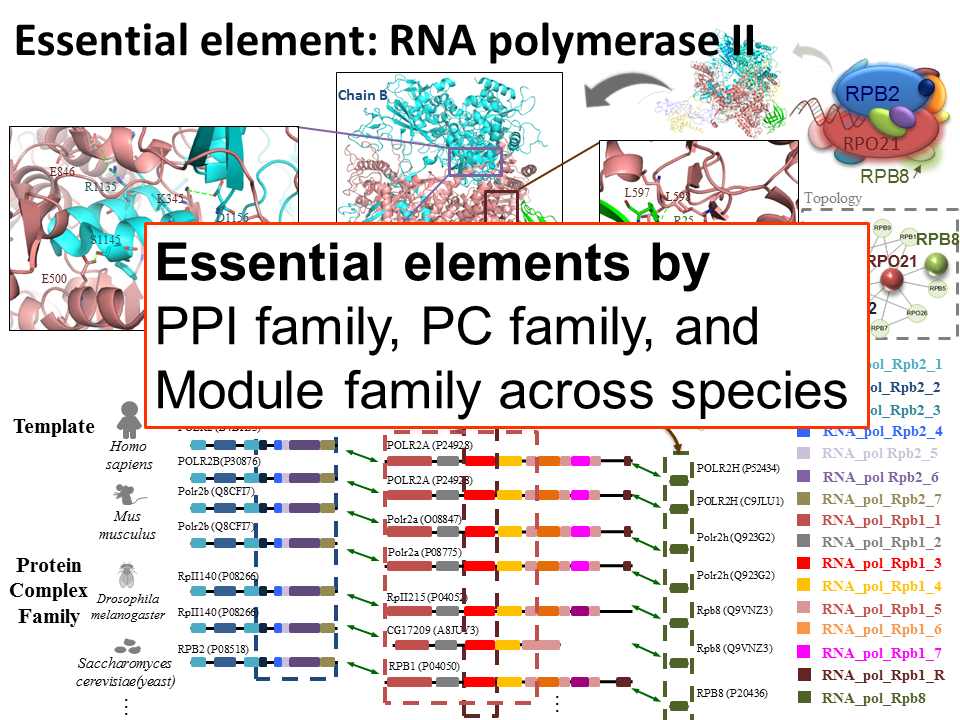
Homologous structural systems biologyFor systems biology and structural bioinformatics, we have proposed a novel concept, called "3D-domain interologs", to discover protein-protein interactions[6-8] and protein-DNA interactions [12] which are considered as orthologous interactions among different species, in particular about inferred interacting domain pairs and binding models (e.g. hydrogen-bond interactions and conserved residues). For protein-protein interactions, we proposed the PPISearch server [5] for searching homologous protein-protein interactions (PPIs), called PPI family, across multiple species. According to our knowledge, PPISearch is the first public server that identifies homologous PPIs from annotated PPI databases and infers transferability of interacting domains and functions between homologous PPIs and the query. By taking account of structural bioinformatics cross species, we extended PPISearch to PCFamily [8], which is useful for binding model visualizations and annotating query proteins based on homologous protein complexes. Until September 2010, over 3,500 users have used PPISearch and PCFamily servers. Furthermore, the number of accessing 3D-BLAST exceeds 6,000 from 44 countries and related papers were cited ~50 times since 2007. |
|
 |
|
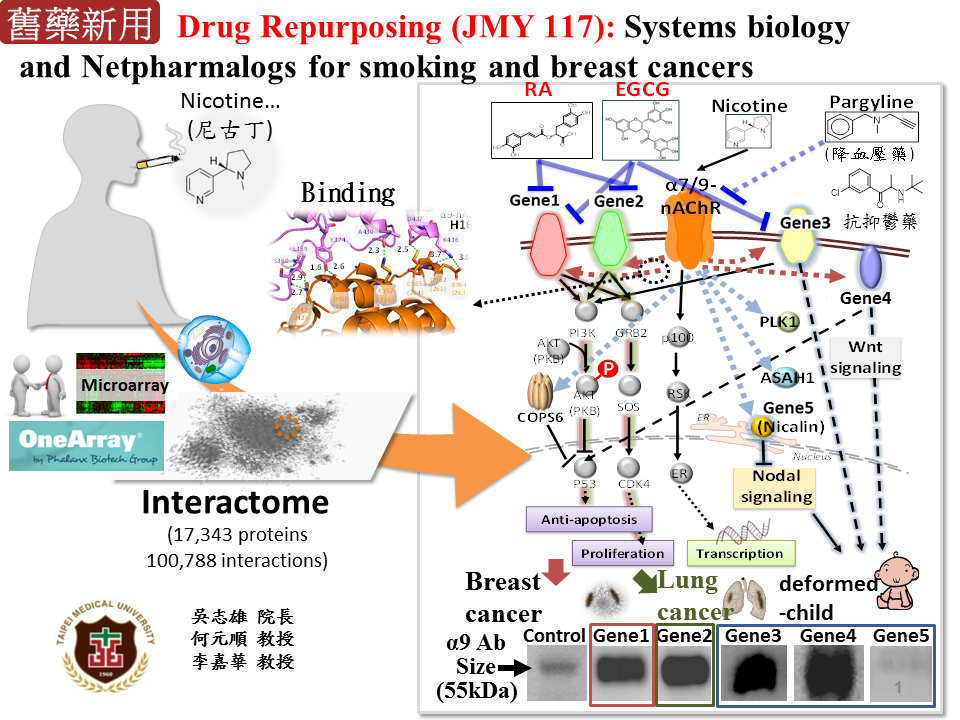
藥物傳輸老藥新用:尼古丁乙醯膽鹼受體α9在尼古丁誘導之癌症中的調控機制本團隊與台北醫學大學乳癌研究團隊(何元順教授團隊及雙和醫院院長 吳志雄教授等)合作,探討尼古丁(Nicotine)誘導多種疾病(如肺癌、頭頸癌、乳癌、畸形兒等)發生之轉譯醫學研究,尼古丁乙醯膽鹼受體α9(α9-nAChR)被認為參與由尼古丁所誘發的人類正常乳腺上皮細胞的轉型,由於目前所有的PPI資料庫都缺乏α9-nAChR的交互作用蛋白質,我們透過同源映射網路及蛋白質-化合物交互作用家族概念,發現5個新的α9-nAChR之交互作用蛋白質(X1、X2、X3、X4和X5)及其相關生化途徑。透過免疫共沉澱及螢光共振能量轉移影像(Förster resonance energy transfer),發現α9-nAChR正常情況下會與生長因子受體(如X1、X2)結合,降低其活性;當尼古丁結合α9-nAChR會使其與生長因子受體分離,因而活化下游生化途徑抑制細胞凋亡及促進細胞增生。目前已將此發現透過臨床檢體進行驗證,成功發現尼古丁誘發乳癌之機制,相關成果發表撰寫中。 |
|
 |
|
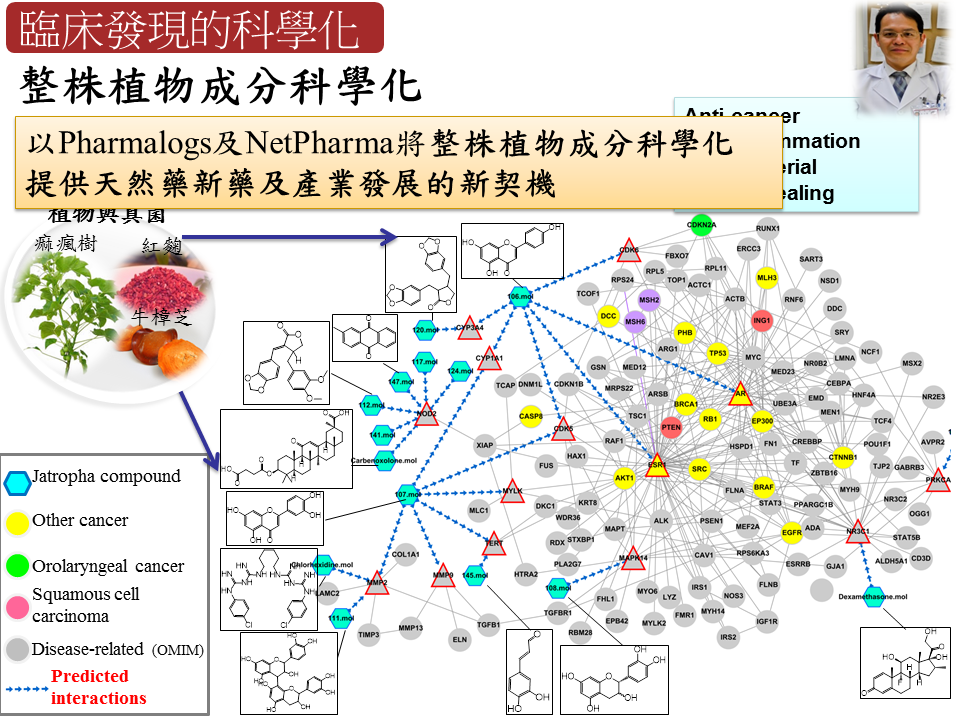
天然藥新藥開發中草藥在中華民族的生活與歷史上扮演重要角色,然而隨著西風東漸,國人因為中草藥的傳統理論基礎缺乏現今科學化的解釋而漸趨勢微,這無論對已有數千年臨床驗證的中草藥或是對人類的健康都是一大損失,所以將中草藥的療效賦予科學化的解釋是相當重要且刻不容緩的。本實驗室透過系統生物的策略,建構出跨物種的同源交互作用網絡,並成功解釋疾病的致病機制。並透過在藥物設計領域的積累,提出全球首創的同源藥理概念,可利用蛋白與藥物的共結晶結構預測出其他具有類似結合機制的標靶蛋白與藥物,有助於舊藥新用與副作用預測。而本實驗室進一步將系統生物與藥物設計結合,可透過將中草藥或整株植物的成分以同源藥理概念找出潛在作用蛋白,並標註於同源交互作用網絡,如此將可以作用標靶、作用途徑、影響疾病等科學化方式闡述中草藥的療效,目前並已成功應用於痲瘋樹、紅麴與牛樟芝的治療與保健功效預測上。 |
|
 |
|
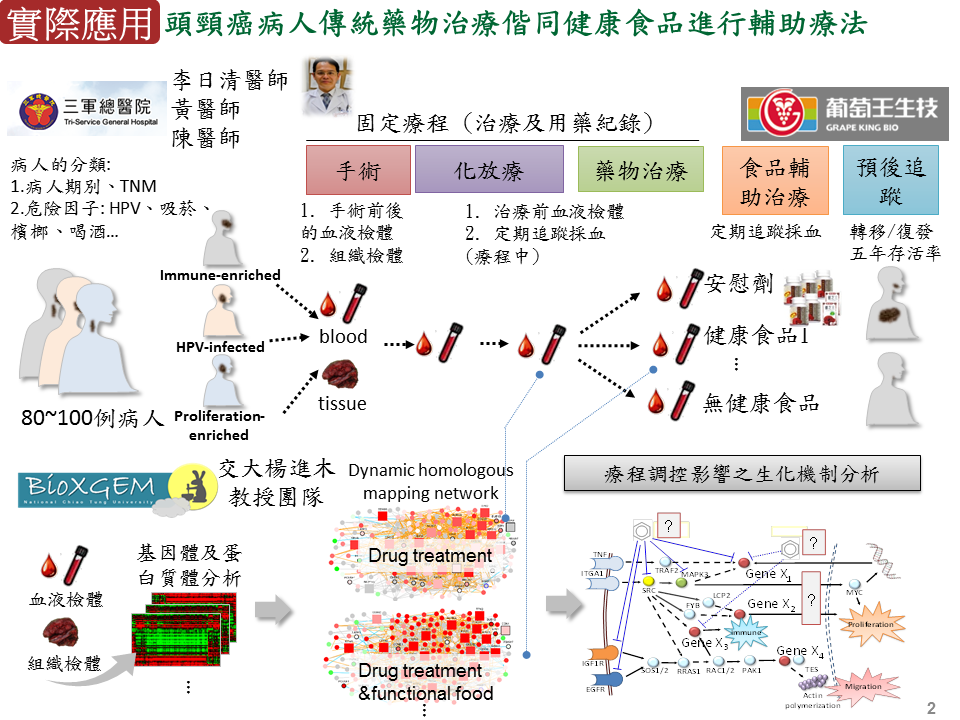
抗癌與抗感染藥物開發:頭頸癌病人傳統藥物治療偕同健康食品進行輔助療法本團隊與三軍總醫院頭頸癌研究團隊(病理部聶鑫主任及李日清醫師)合作,透過系統生物策略模型,發展可以用於診斷、預後及治療之頭頸癌生物標記-HNC88(88個基因),並試圖以 HNC88 從分子階層角度對病患進行分類與治療,大致可將病患分成 HPV-infected、Immune-enriched 及 Proliferation-enriched 三大類。葡萄王生技為國內保健食品製造龍頭,本團隊與葡萄王生技合作進行生技健康食品科學化及功效驗證,如:分析樟芝王關鍵成分對口腔癌病患化放療之副作用緩解分析及預後評估,探討食用前、後之各病患子類血液中基因表現與癌症生化途徑的變化。 |
|
 |
|
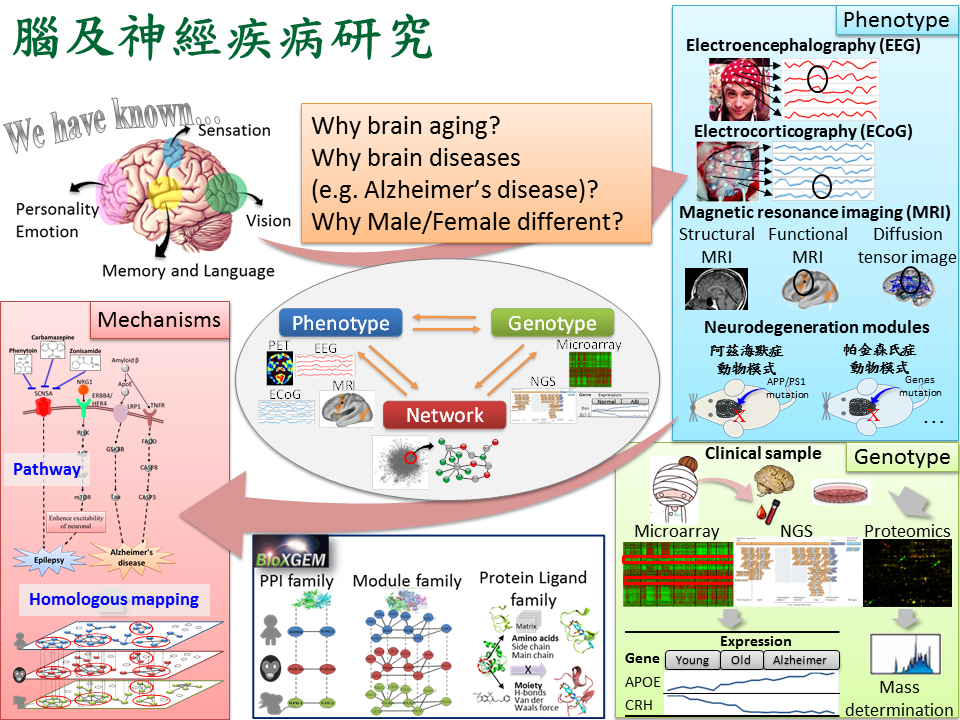
腦及神經老化藥物開發人類對於大腦運作所負責的腦區已有了解但對於詳細運作機制仍然尚未釐清,因此對於大腦研究進而衍伸出幾個議題:1. 因應世界人口老齡化的趨勢因而衍生出老年神經退化性疾病,例如:阿茲海默症,根據統計在 2010 年全球有將近 2100 萬到 3500 萬名阿茲海默症患者,而歸因於阿茲海默症相關的死亡案例大約有 48.6 萬例。阿茲海默症會使患者越來越需要他人的照護,因此導致在已開發國家中阿茲海默症是相當耗費社會資源的疾病之一。世界衛生組織指出目前並沒有特定藥物有實證證明對阿茲海默症治療有效,只有少數可能可以暫時(緩解)改善症狀的方法,而健康食品及植物藥被視為是另類可能的有效途徑;2. 由於許多大腦疾病已知與性別有關,例如:男生易患自閉症、女生易患阿茲海默症,因此透過了解男女大腦先天上的差異對於釐清疾病病理機制具有莫大幫助,本團隊透過表徵型資料:腦電波圖譜(EEG)、大腦核磁共振(MRI)、動物模式,以及基因型資料:基因表達圖譜(mRNA microarray)、次世代定序(NGS)、蛋白質體圖譜(Proteomics),整合本團隊核心技術蛋白質交互作用家族(PPI family)、模組交互作用家族(Module family)、蛋白質-配體交互作用家族(Protein-ligand family),進而揭露大腦疾病詳細生化機制。 |
|
 |
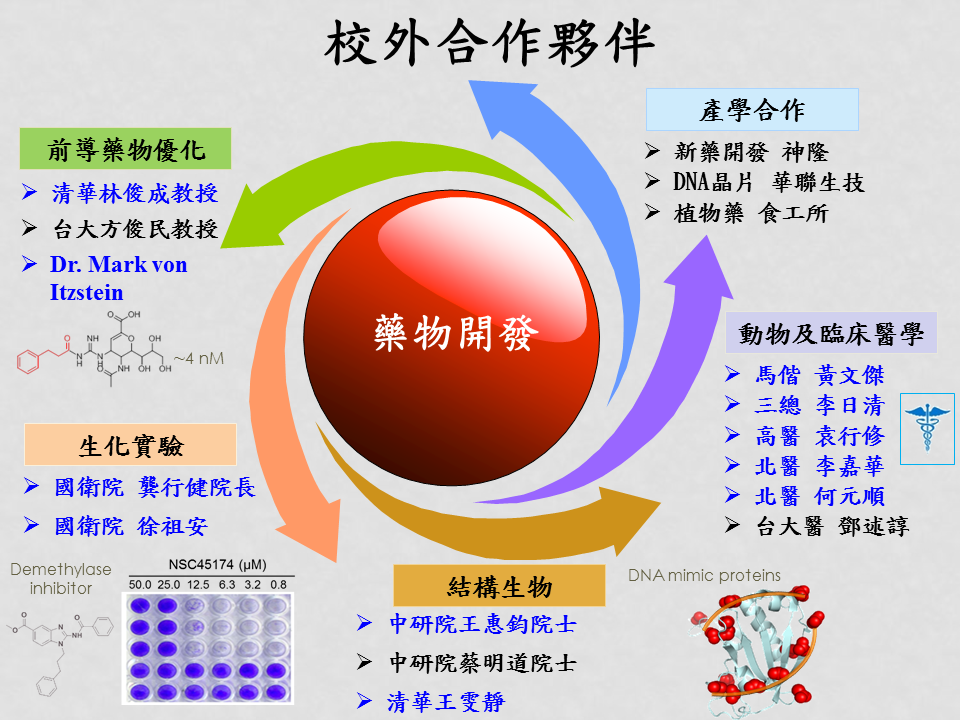 |
合作夥伴We have accumulated extensive experiences in drug design to achieve the specific aims. Our team consists of experts in different fields, including computational drug design, systems biology, clinical trial, lead optimization, biochemistry, and animal models. We have successfully discovered 45 lead compounds (<10 μM) for various diseases or pathogens. One of the identified natural compounds, rosmarinic acid, is currently applying for the U.S. patent and IRB approval at Mackay Memorial Hospital (馬偕醫院) for clinical trials. Based on these results, we have received one U.S. patent (US 8,175,860 B2), and have applied one U.S. patent (13/872,456) and one ROC patent. In university-industry collaborations, we have collaborated with ScinoPharm (台灣神隆), Phalanx Biotech Group (華聯生技), Food Industry Research and Development Institute (食品工業研究所), and Jason Life Tech for developing new drugs, botanical drugs, healthy food, and microarray platform for detecting diseases. We have collaborated with over 30 laboratories (e.g., Professor Wang HJ [王惠鈞院士] and Professor Mark von Itzstein, inventor of the first anti-influenza drug, Zanamivir), five teaching hospitals, and 15 national institutes for drug discovery, systems biology, binding mechanisms, and multi-target drugs. In addition, we have achieved some significant results in molecular interactions and structural systems biology: 1) We are the first team proposed molecular-interface families; 2) We have established the statistical biophysics methods to infer site-moiety maps (hotspots) of proteins for protein-ligand binding mechanisms and drug discovery; 3) We have proposed the comprehensive molecular interaction networks for studying interactomes; 4) We have developed GEMDOCK, which is a world-wide used docking programs. |
|
References
- Chen YF, Hsu KC, Lin SR, Wang WC, Huang YC, Yang JM: SiMMap: a web server for inferring site-moiety map to recognize interaction preferences between protein pockets and compound moieties. Nucleic Acids Research 2010, 38:W424-W430.
- Yang JM, Chen YF, Tu YY, Yen KR, Yang YL: Combinatorial computational approaches to identify tetracycline derivatives as flavivirus inhibitors. PLoS ONE 2007:e428.
- Yang JM, Chen CC: GEMDOCK: a generic evolutionary method for molecular docking. Proteins: Structure, Function, and Bioinformatics 2004, 55:288-304.
- Yang JM, Shen TW: A pharmacophore-based evolutionary approach for screening selective estrogen receptor modulators. Proteins: Structure, Function, and Bioinformatics 2005, 59:205-220.
- Chen CC, Lin CY, Lo YS, Yang JM: PPISearch: a web server for searching homologous protein-protein interactions across multiple species. Nucleic Acids Research 2009, 37:W369-W375.
- Chen YC, Lo YS, Hsu WC, Yang JM: 3D-partner: a web server to infer interacting partners and binding models. Nucleic Acids Research 2007:W561-W567.
- Lo YS, Chen YC, Yang JM: 3D-interologs: An evolution database of physical protein-protein interactions across multiple genomes. BMC Genomics 2010, 11, Suppl 3:S7.
- Lo YS, Lin CY, Yang JM: PCFamily: a web server for searching homologous protein complexes. Nucleic Acids Research 2010, 38:W516-W522.
- Yang JM, Tung CH: Protein structure database search and evolutionary classification. Nucleic Acids Research 2006, 34:3646-3659.
- Tung CH, Huang JW, Yang JM: Kappa-alpha plot derived structural alphabet and BLOSUM-like substitution matrix for rapid search of protein structure database. Genome Biology 2010, 2007, 8:R31.
- Tung CH, Yang JM: fastSCOP: a fast web server for recognizing protein structural domains and SCOP superfamilies. Nucleic Acids Research 2007, 35:W438-W443.
- Chang YL, Tsai HK, Kao CY, Chen YC, Hu YJ, Yang JM: Evolutionary conservation of DNA-contact residues in DNA-binding domains. BMC Bioinformatics 2008, 9(S6):S3.1-S3.9.
- Yang JM, Chen YF, Shen TW, Kristal BS, Hsu DF: Consensus scoring criteria for improving enrichment in virtual screening. Journal of Chemical Information and Modeling 2005, 45:1134-1146.
- Yang JM: Development and evaluation of a generic evolutionary method for protein-ligand docking. Journal of Computational Chemistry 2004, 25:843-857.
- Hung HC, Tseng CP, Yang JM, Ju YW, Tseng SN, Chen YF, Chao YS, Hsieh HP, Shih SR, Hsu JT: Aurintricarboxylic acid inhibits influenza virus neuraminidase. Antiviral Research 2009, 81:123-131.
- Yang MC, Guan HH, Liu MY, Lin YH, Yang JM, Chen WL, Chen CJ, Mao SJT: Crystal Structure of a Secondary Vitamin D3 Binding Site of Milk ß-Lactoglobulin. Proteins: Structure, Function, and Bioinformatics 2008:1197-1210.
- Yang MC, Guan HH, Yang JM, Ko CN, Liu MY, Lin YH, Chen CJ, Mao SJT: Rational Design for crystallization of b-Lactoglobulin and vitamin D3 complex: reveal of a secondary binding site. Crystal Growth & Design 2008:DOI: 10.1021/cg800697s.
- Chin KH, Lee YC, Tu ZL, Chen CH, Tseng YH, Yang JM, Ryan RP, McCarthy Y, Dow JM, Wang AH et al: The cAMP receptor-like protein CLP is a novel c-di-GMP receptor linking cell-cell signaling to virulence gene expression in Xanthomonas campestris. Journal of Molecular Biology 2010, 396:646-662.
工具及資料庫
BioXGEM 所發展的生物資訊網頁工具及資料庫
GEMDOCK
GEMDOCK - a Generic Evolutionary Method for molecular DOCKing GEMDOCK is a program for computing a ligand conformation and orientation relative to the active site of target protein. The tool was developed by Jinn-Moon Yang, a profesor of the Institute of Bioinformatics, National Chiao Tung University.
URL: http://gemdock.life.nctu.edu.tw/dock/
SiMMap
SiMMap: Site-moiety map for drug discovery and mechanisms
URL: http://simfam.life.nctu.edu.tw/
3D-BLAST
3D-BLAST is a very fast and accurate method for discovering the homologous proteins and evolutionary classifications of a newly determined protein structure. Our 3D-BLAST has the advantages of BLAST tool for fast protein structure database scanning.
URL: http://3d-blast.life.nctu.edu.tw/
Protein-Protein Interaction Search
Input an interacting protein pair as a query to search its homologous interactions across multiple species
URL: http://gemdock.life.nctu.edu.tw/ppisearch/
PCFamily
PCFamily : Protein Complex Family Search
URL: http://pcfamily.life.nctu.edu.tw/
MoNetFamily
MoNetFamily - Homologous modules and module-module interaction networks in vertebrates
URL: http://monetfamily.life.nctu.edu.tw/
KIDFamMap
KIDFamMap: Kinase-inhibitor-disease family map
URL: http://gemdock.life.nctu.edu.tw/KIDFamMap/
CaMPNets
CaMPNets: Membrane protein-regulated networks across human cancers
URL: http://campnets.life.nctu.edu.tw/
PharmaCoNets
PharmaCoNets: Drug community–regulated networks
URL: http://pharmaconets.life.nctu.edu.tw/
BioXGEM Integrated Discovery for Omics data
URL: http://140.113.120.248/BioXGEM-tools/BioXGEM Integrated Discovery for Drug Design
URL: http://140.113.120.248/BioXGEM-drug/FooDisNET
URL: http://140.113.120.248/FooDisNETTranditional Chinese Herb
Tranditional Chinese Herb
URL: http://gemdock.life.nctu.edu.tw/HERBID/
BioXGEM 常用 Data Sets
SARS-CoV-2 main protease data (Download all)
Molecular recognition
Virtual screening sets
Gene expression sets
Breast cancer
Liver cancer
Head and neck cancer
Diabetes
Convolutional neural network for human cancer types prediction
常用連結
Useful Links for Paper (Thesis) Writing and Research Career Advice
- How to be a Good Graduate Student/Advisor, by Marie desJardins
- You and Your Research [English.pdf] [Chinese.pdf] by Richard W. Hamming
- Useful Things To Know about Ph.D. Thesis Research by H. T. Kung
- Better Dissertation Writing in English from Prof. Yuh-Dauh Lyuu's homepage
- How to Organize your Thesis, by John W. Chinneck
- Writing and Presenting Your Thesis or Dissertation, by S. Joseph Levine, Ph.D.
- The Elements of Style, 4th ed. by William Strunk, Jr., E.B. White, and Roger Angell. The 1918 version (free).
- The Chicago Manual of Style
- Research career advice from David Patternson's homepage
Protein
| PDB | This resource is powered by the Protein Data Bank archive-information about the 3D shapes of proteins, nucleic acids, and complex assemblies that helps students and researchers understand all aspects of biomedicine and agriculture, from protein synthesis to health and disease. |
| UniProt | The mission of UniProt is to provide the scientific community with a comprehensive, high-quality and freely accessible resource of protein sequence and functional information. |
| PDBsum | The PDBsum is a pictorial database that provides an at-a-glance overview of the contents of each 3D structure deposited in the Protein Data Bank (PDB). It shows the molecule(s) that make up the structure (ie protein chains, DNA, ligands and metal ions) and schematic diagrams of their interactions. Extensive use is made of the freely available RasMol molecular graphics program to view the molecules and their interactions in 3D. |
| CATH | CATH is a classification of protein structures downloaded from the Protein Data Bank. We group protein domains into superfamilies when there is sufficient evidence they have diverged from a common ancestor. |
| PFam | The Pfam database is a large collection of protein families, each represented by multiple sequence alignments and hidden Markov models (HMMs). Proteins are generally composed of one or more functional regions, commonly termed domains. Different combinations of domains give rise to the diverse range of proteins found in nature. The identification of domains that occur within proteins can therefore provide insights into their function. |
| Dali Database | The Dali Database is based on all-against-all 3D structure comparison of protein structures in the Protein Data Bank (PDB). The structural neighbourhoods and alignments are automatically maintained and regularly updated using the Dali search engine. |
| SWISS-MODEL | SWISS-MODEL is a fully automated protein structure homology-modelling server. |
| (PS)2 | (PS)2 is an automatic homology modeling server. |
| The ConSurf Server | It provides evolutionary conservation profiles for proteins of known structure in the PDB. |
| THPA | The Human Protein Atlas portal is a publicly available database with millions of high-resolution images showing the spatial distribution of proteins in 44 different normal human tissues and 20 different cancer types, as well as 46 different human cell lines. |
| STRING | STRING is a database of known and predicted protein interactions. The interactions include direct (physical) and indirect (functional) associations; they are derived from four sources: Genomic Context, High-throughput Experiments, (Conserved) Coexpression and Previous Knowledge. |
| Brenda | The comprehensive enzyme information system |
| PhosphoSitePlus | PhosphoSitePlus® (PSP) is an online systems biology resource providing comprehensive information and tools for the study of protein post-translational modifications (PTMs) including phosphorylation, ubiquitination, acetylation and methylation. |
| BLAST | For blast search to find similar DNA or Protein sequences |
| CLUSTAL Omega and other MSA tools | For Protein MSA |
| T-Coffee | T-Coffee is a multiple sequence alignment program. Its main characteristic is that it will allow you to combine results obtained with several alignment methods. |
| WebLogo | WebLogo is a web based application designed to make the generation of sequence logos as easy and painless as possible. |
| WebLogo 3 | WebLogo is a web based application designed to make the generation of sequence logos easy and painless. |
| Back to top |
Compound
| PubChem | PubChem is a database of chemical molecules and their activities against biological assays. |
| ChEMBL | It is a manually curated chemical database of bioactive molecules with drug-like properties. |
| DrugBank | The DrugBank database is a unique bioinformatics and cheminformatics resource that combines detailed drug. |
| BindingDB | BindingDB is a public, web-accessible database of measured binding affinities, focusing chiefly on the interactions of protein considered to be drug-targets with small, drug-like molecules. |
| STITCH | STITCH is a resource to explore known and predicted interactions of chemicals and proteins. |
| Avogadro | Molecule building and editing tool to build and minimize compound structures |
| TCMID | Provide information on all respects of Traditional Chinese Medicine including formulae, herbs and herbal ingredients |
| Dr. Duke's Phytochemical and Ethnobotanical Databases | Provide the infomation about plants and their ingredients. |
| EDB | Provide the infomation about plants in Bangladesh and their ingredients. |
| ZINC | A free database of commercially-available compounds. |
| MolPort | MolPort is advanced chemical marketplace. |
| Selleckchem | Selleck Chemicals supplies over 3,000 inhibitors used in the study of cell signaling pathways. We actively tracks the latest science so our customers can rely on us to be the leading supplier of the newest inhibitors. |
| Chemical Book | 查詢化合物相關資訊的網站 |
| Back to top |
Gene
| Gene Ontology | The Gene Ontology (GO) project is a major bioinformatics initiative to develop a computational representation of our evolving knowledge of how genes encode biological functions at the molecular, cellular and tissue system levels. |
| Gene Expression Data (L1000) | Gene expression is highly correlated. We take advantage of this high degree of correlation to reduce the number of measurements needed to generate meaningful gene expression data for the approximately 20,000 genes in the human genome. |
| DAVID | DAVID now provides a comprehensive set of functional annotation tools for investigators to understand biological meaning behind large list of genes. |
| GEO (Gene Expression Omnibus) | GEO is a public functional genomics data repository supporting MIAME-compliant data submissions. Array- and sequence-based data are accepted. Tools are provided to help users query and download experiments and curated gene expression profiles. |
| Genome | This resource organizes information on genomes including sequences, maps, chromosomes, assemblies, and annotations. |
| 1000 Genome Browser | A global reference for human genetic variation |
| TCGA (The Cancer Genome Atlas) | The Cancer Genome Atlas (TCGA) Data Portal provides a platform for researchers to search, download, and analyze data sets generated by TCGA. It contains clinical information, genomic characterization data, and high level sequence analysis of the tumor genomes. |
| cBioPortal | The cBioPortal for Cancer Genomics provides visualization, analysis and download of large-scale cancer genomics data sets (e.g. TCGA). |
| CCLE (Cancer Cell Line Encyclopedia) | The Cancer Cell Line Encyclopedia (CCLE) project is an effort to conduct a detailed genetic characterization of a large panel of human cancer cell lines. The CCLE provides public access analysis and visualization of DNA copy number, mRNA expression, mutation data and more, for 1000 cancer cell lines. |
| GeneCards | GeneCards is a searchable, integrative database that provides comprehensive, user-friendly information on all annotated and predicted human genes. It automatically integrates gene-centric data from ~125 web sources, including genomic, transcriptomic, proteomic, genetic, clinical and functional information. |
| HPRD | Provide integrated information of domain architecture, post-translational modifications, interaction networks and disease association for each protein in the human proteome. |
| Allen Brain Atlas | An anatomically comprehensive atlas of the adult human brain transcriptome |
| MGI | Provide integrated genetic, genomic, and biological data for mouse. |
| SGD | Provide integrated biological information for the budding yeast Saccharomyces cerevisiae. |
| ArrayExpress | ArrayExpress Archive of Functional Genomics Data stores data from high-throughput functional genomics experiments, and provides these data for reuse to the research community. |
| Back to top |
Tools
| Chimera | Protein Visualization tool |
| Cytoscape | Cytoscape is an open source software platform for visualizing complex networks and integrating these with any type of attribute data. |
| MeV | MeV is a versatile microarray data analysis tool, incorporating sophisticated algorithms for clustering, visualization, classification, statistical analysis and biological theme discovery. |
| GENE-E | GENE-E is a matrix visualization and analysis platform designed to support visual data exploration. |
| Back to top |
Others
| Google 學術搜尋 | 站在巨人的肩膀上 |
| KEGG | KEGG PATHWAY is a collection of manually drawn pathway maps representing our knowledge on the molecular interaction and reaction networks. |
| EBI (The European Bioinformatics Institute) | EMBL-EBI provides freely available data from life science experiments, performs basic research in computational biology and offers an extensive user training programme, supporting researchers in academia and industry. |
| COSMIC | All cancers arise as a result of the acquisition of a series of fixed DNA sequence abnormalities, mutations, many of which ultimately confer a growth advantage upon the cells in which they have occurred. There is a vast amount of information available in the published scientific literature about these changes. COSMIC is designed to store and display somatic mutation information and related details and contains information relating to human cancers. |
| Ensembl | Including Gene expression database, SNP database and ENCODE (Encyclopedia of DNA Elements (ENCODE) Consortium) *The Encyclopedia of DNA Elements (ENCODE) Consortium is an international collaboration of research groups funded by the National Human Genome Research Institute (NHGRI) |
| RefSeq: NCBI Reference Sequence Database | A comprehensive, integrated, non-redundant, well-annotated set of reference sequences including genomic, transcript, and protein. |
| ExPASy | ExPASy is the SIB Bioinformatics Resource Portal which provides access to scientific databases and software tools (i.e., resources) in different areas of life sciences including proteomics, genomics, phylogeny, systems biology, population genetics, transcriptomics etc. On this portal you find resources from many different SIB groups as well as external institutions. |
| SMART | SMART (a Simple Modular Architecture Research Tool) allows the identification and annotation of genetically mobile domains and the analysis of domain architectures. More than 500 domain families found in signalling, extracellular and chromatin-associated proteins are detectable. These domains are extensively annotated with respect to phyletic distributions, functional class, tertiary structures and functionally important residues. Each domain found in a non-redundant protein database as well as search parameters and taxonomic information are stored in a relational database system. User interfaces to this database allow searches for proteins containing specific combinations of domains in defined taxa. For all the details, please refer to the publications on SMART. |
| BioPharma Insight | BioPharm Insight is your definitive guide to the global life sciences community, combining an online business intelligence system of comprehensive market analytics and key industry contacts with an independent investigative journalism news service. |
| GlobalData | GlobalData is the premier source of actionable insight into the energy and healthcare industries. With the combined expertise of more than 1,000 researchers, market analysts and consultants, we provide high-quality, accurate and transparent industry insight that helps our clients to achieve growth and increase business value. |
| GEN | Genetic Engineering & Biotechnology News (GEN) has retained its position as the premier biotech publication since its launch in 1981. GEN publishes a print edition 21 times a year and has additional exclusive editorial content online, including news and analysis as well as webinars, videos, and polls. GEN's unique news and technology focus covers the entire bioproduct life cycle, including drug discovery, early-stage R&D, applied research (e.g., omics, biomarkers, and diagnostics), bioprocessing, and commercialization. |
| Phys Org: Biology news | |
| 海峽生技醫藥品風雲 | |
| Back to top |
人才招募
| 歡迎有熱情 有想法的碩博生 專題生加入我們的行列 | ||||
| 誠徵博士後研究員 碩士級專任研究助理 | ||||
| THE WORLD OF DRUG DISCOVERY, | ||||
| FUNCTION BIOINFORMATICS AND SYSTEMS BIOLOGY. | ||||
|
楊進木 教授 03-5712121 #56942 (Lab: #56946) moon@nycu.edu.tw 賢齊館 415-1 室 |
 |
|||
研究內容
|
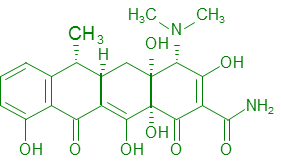
發現四環抗生素對 |
|||
|
|

|
||
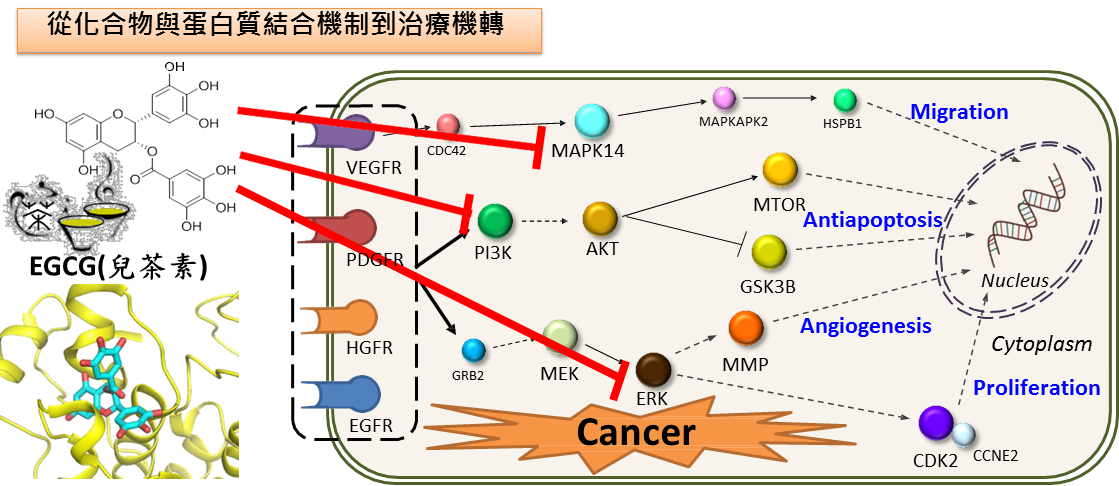
|
||||
 |
 |
 |
活動花絮
活動照片
運動
運動
時間 ► 2015年5月7日 (星期四)
地點 ► 國立交通大學光復校區 排球場
參加生科系運排球比賽賽後合照

出遊
時間 ► 2014年2月18日 (星期二)
地點 ► 老師的農莊
在老師的農莊摘橘子



耶誕晚會
時間 ► 2013年12月23日 (星期一)
地點 ► 國立交通大學博愛校區 生科實驗館 生資中心
耶誕晚會交換禮物






謝師宴
時間 ► 2013年8月7日 (星期三)
地點 ► 新竹加賀日本料理
在加賀日本料理舉辦謝師宴



成員合照




研討會集錦




